Neonatal duodenal atresia with heterotopic pancreas: a case report and literature review
Yinmin Sun, Siqi Li, Shiqi Liu, Yufeng Li, Dongwen Quan

TL;DR
A neonate with duodenal atresia was found to have heterotopic pancreas, leading to a recommendation for screening and early removal to prevent future complications.
Contribution
This case report highlights the novel association between duodenal atresia and heterotopic pancreas, advocating for systematic screening and concurrent resection.
Findings
Heterotopic pancreas was found in a neonate with duodenal atresia during surgery.
The neonate remained stable post-surgery with no recurrence after 12 months.
The authors recommend screening for heterotopic pancreas in neonates with duodenal atresia.
Abstract
Heterotopic pancreas (HP) is a congenital anomaly characterized by pancreatic tissue entirely separate from the orthotopic pancreas, lacking direct ductal communication and vascular continuity. HP is most frequently found in the upper gastrointestinal tract, particularly the stomach, duodenum, and proximal jejunum, while involvement of the mesentery is relatively rare We report a case of congenital duodenal atresia with concomitant HP in a 3-day-old neonate. Emergency laparotomy was performed at 72 h of life for membranous duodenal atresia; web excision and duodenoduodenostomy were completed. Preoperative evaluation showed no other associated malformations (e.g., congenital heart disease or Down syndrome). Intraoperatively, a 10.0 × 6.0 mm focus of HP tissue was unexpectedly found at the jejunal mesenteric boundary, approximately 5–7 cm distal to the duodenal obstruction, necessitating…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
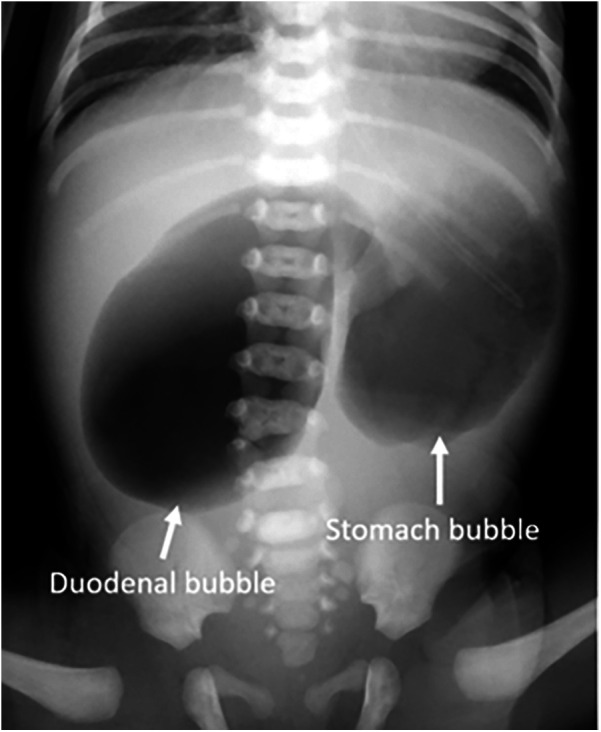 Figure 1
Figure 1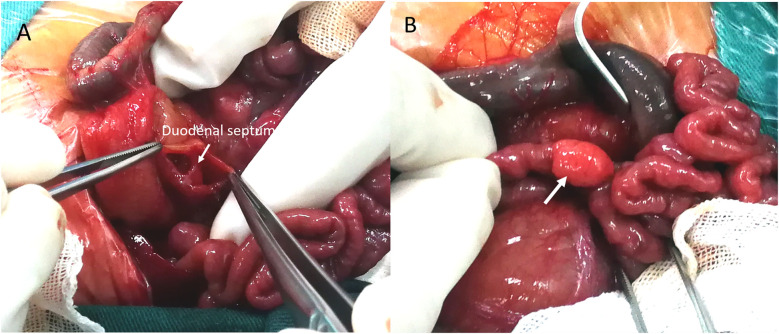 Figure 2
Figure 2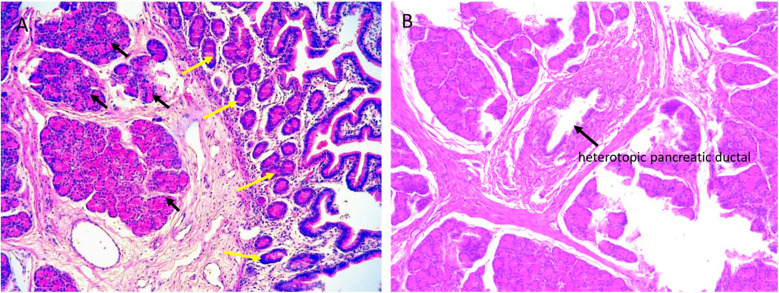 Figure 3
Figure 3| Source. | From | No. | Age | Sex | Location | Heinrich type | Methods of diagnosis | Clinical manifestation | Resection | Follow up |
|---|---|---|---|---|---|---|---|---|---|---|
| Koltai ( | German | 1 | 1.5 yr | F | Mesocolon | N/A | US + HP | Loss of appetite + right mid-abdominal mass | Yes | N/A |
| Fam ( | America | 1 | 12 yr | M | Jejunal mesentery | N/A | surgical exploration + HP | Abdominal pain around the umbilicus + nausea + vomiting | Yes | N/A |
| Iuchtman ( | Israel | 1 | 35 yr | F | Mesocolon | N/A | US + HP | Cystic lesions incidentally detected on ultrasound | Yes | N/A |
| Tornóczky ( | Hungary | 1 | 15 yr | F | Mesocolon | Tumor | CT + HP | Abdominal pain + bloating | Yes | N/A |
| Matsumoto ( | Japan | 1 | 3 yr | F | Mesocolon | N/A | US + CECT + HP | abdominal pain | Yes | 20 months, asymptomatic |
| Silva ( | America | 1 | 57 yr | F | Mesentery | N/A | CECT + MRCP | Right back pain + full abdominal pressure + nausea | No | N/A |
| Shin ( | Korea | 1 | 38 yr | M | Jejunal mesentery | N/A | CECT + HP | One syncopal episode + black stools + no pressure in abdomen | Yes | 9 months, no recurrence |
| Canbaz ( | Turkish | 1 | 75 yr | F | Jejunal mesentery | N/A | US + HP | Periumbilical pain, nausea, vomiting + elevated amylase and lipase | Yes | 1year, no recurrence |
| Yang ( | China | 1 | 7 yr | F | Mesentery | Pancreatic blastoma | US + CT + MRI + HP | Abdominal pain + vomiting | Yes | 3 years, no recurrence of tumor |
| Wong ( | Canadian | 1 | 67 yr | F | Jejunal mesentery | N/A | CECT + MRCP + HP | Epigastric tingling radiating to the back + Nausea + Vomiting + Elevated serum lipase | Yes | 6 months, no recurrence |
| Wang ( | China | 1 | 3mo | F | Jejunal mesentery | N/A | Intraoperative Finding + HP | Asymptomatic + incidental intraoperative findings | N/A | N/A |
| Vyas ( | India | 1 | 19 yr | M | Mesentery | N/A | CECT + MRI | Abdominal pain + fever + easy fatigue | N/A | N/A |
| Ginsburg ( | America | 1 | 15 yr | F | Jejunal mesentery | I | CECT + HP | Total abdominal pain + elevated serum lipase and amylase | Yes | N/A |
| Seo ( | Korea | 7 | Avg 50 yr (range 36–61) | 3M/4F | Jejunal mesentery | N/A | CECT | Other diseases found on CT examination | No | 26–77 months (median 39 months), asymptomatic |
| Borghol ( | French | 1 | 65 yr | M | Jejunal mesentery | N/A | CT | Asymptomatic + follow-up imaging for colon cancer | No | N/A |
| Navickas ( | Lithuania | 1 | 49 yr | M | Jejunal mesentery | I | CECT + HP | Asymptomatic + excludes prostate cancer metastasis incidental findings | Yes | N/A |
| Moreno ( | Netherlands | 1 | 58 yr | F | Jejunal mesentery | I | CECT + MRI + EUS - FNA + HP | Acute abdominal pain + nausea + vomiting | Yes | N/A |
| De ( | Japan | 1 | 55 yr | F | Jejunal mesentery | N/A | CT + MRCP + HP | Upper abdominal pain + elevated blood amylase and lipase | Yes | N/A |
| Tang ( | Spanish | 1 | 59 yr | F | Jejunal mesentery | N/A | MDCT + EUS- FNA + MRCP | Upper abdominal pain + elevated lipase + elevated CA19.9 | No | 1 month, Ca 19.9 and lipase normalized |
| Yamakawa ( | China | 1 | 12 yr | F | Jejunal mesentery | I | US + CECT | Abdominal pain + peritoneal irritation + intermittent vomiting | Yes | 1 year, no recurrence |
| Aslan ( | Turkish | 1 | 44 yr | F | Jejunal mesentery | N/A | CECT | abdominal pain | No | 2 years,no signs of malignancy |
| Xu ( | African | 1 | 7 yr | M | Jejunal mesentery | I | HP | Right lower abdominal pain + non-bilious vomiting | Yes | 3 months, no recurrence |
| Sathiadoss ( | Canadian | 1 | 45 yr | F | Jejunal mesentery | II | CECT + HP | Nausea, non-bilious vomiting and mild epigastric pain (lasting 4 years) | Yes | N/A |
| Bullock ( | America | 1 | 43 yr | F | Jejunal mesentery | I | CECT + MRI + MRCP | Frequent abdominal pain + elevated lipase | Yes | N/A |
| Okamura ( | Japan | 1 | 61 yr | M | Jejunal mesentery | II | CECT + MRCP | Abdominal pain + 4 history of pancreatitis | Yes | 2 years, no recurrence |
| Our case | China | 1 | 3 d | F | Jejunal mesentery | I | Intraoperative Finding + HP | Asymptomatic + incidental intraoperative findings | Yes | 1 year, asymptomatic |
- —National Natural Science Foundation of China
- —Natural Science Foundation of Shaanxi Provincial Key Industries Innovation Chain (Cluster)-Social Development Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIntestinal Malrotation and Obstruction Disorders · Gastrointestinal disorders and treatments · Pediatric Hepatobiliary Diseases and Treatments
Introduction
1
Congenital duodenal obstruction (CDO), occurring in approximately 1.5 per 10,000 live births, requires early surgical intervention. The co-occurrence of CDO with heterotopic pancreas (HP) represents an exceptionally uncommon clinical presentation. HP refers to pancreatic tissue located outside the native gland, lacking vascular or ductal continuity with the orthotopic pancreas (1). Most individuals with HP remain asymptomatic during life, making its true incidence difficult to determine. Autopsy studies report a prevalence of 0.55% to 15% (1–3), while intraoperative and endoscopic detection rates are approximately 0.18% to 0.2% (4). Due to its frequent subclinical course, HP is often identified incidentally during histologic examination. Reported HP distribution varies across studies due to the absence of systematic evaluation; however, available literature indicates that HP primarily occurs in the stomach, duodenum, jejunum, and Meckel's diverticulum (1, 4). Extraintestinal occurrences (e.g., mesentery, greater omentum, spleen) are significantly rarer, with only isolated cases reported (3, 4).
Herein, we detail a rare case of congenital duodenal atresia associated with jejunal mesenteric heterotopic pancreas (MHP). The neonate required emergency laparotomy 72 h post-birth for CDO. Intraoperatively, HP tissue was incidentally identified at the boundary between the jejunum and its mesentery.
Case description
2
A female 35 + 5-week preterm infant was delivered vaginally following premature rupture of membranes. Antenatal ultrasonography revealed gastric and duodenal dilation with the characteristic “double-bubble sign” and polyhydramnios, prompting admission for suspected CDO.
The mother attended regular prenatal visits. At 32 weeks' gestation, ultrasound demonstrated polyhydramnios and a double-bubble sign, suggesting possible CDO. With no other anomalies detected, she continued routine monitoring. At 35 ^+^ ^5^ weeks, premature membrane rupture resulted in vaginal delivery of a 2,300 g female infant. Apgar scores were 9, 10, and 10 at (1/5/10 min). After birth, the newborn was assessed, with unremarkable findings on evaluation of vital signs, mental status, and physical abdominal examination. At 12 h after delivery, a trial feeding under close monitoring was initiated, beginning with clear water followed by breastfeeding. After increasing the volume of breast milk, the infant developed mild milk vomiting approximately 2 h later. The frequency of emesis increased progressively, and the vomitus transitioned to bilious yellow-green fluid without a feculent odor (5–10 ml/non-projectile episode). Given the prenatal ultrasound findings, radiography was promptly performed. The infant did not pass meconium within the first 24 h and had only two urinations (∼5 ml each), which were without hematuria or mucus. Clinical assessment revealed afebrile status, alertness, and no lethargy/irritability.
Admission laboratory investigations demonstrated the following hematologic and biochemical profiles: white blood cell count (WBC) 16.5 × 10⁹/L (55% neutrophils, 30% lymphocytes); red blood cell count (RBC) 5.2 × 10^12^/L; hemoglobin (Hb) 170.0 g/L; and platelets (PLT) 250.0 × 10⁹/L. Serum electrolytes were sodium (Na^+^) 138.0 mmol/L, potassium (K^+^) 4.0 mmol/L, and chloride (Cl^−^) 98.0 mmol/L. Preoperative serum amylase was 78.0 U/L (reference range: 25–125 U/L) and lipase was 55.0 U/L (reference range: 0–190 U/L). Hepatic and renal function panels were within normal limits. Abdominal radiography revealed marked gastric and duodenal dilation with the classic “double-bubble” sign (Figure 1). These radiographic findings, in conjunction with the clinical presentation, were highly suggestive of congenital duodenal atresia. Preoperative examinations revealed no other associated malformations (e.g., congenital heart disease or Down syndrome) in the neonate.
Preoperative x-ray shows the “double bubble” sign with no gas in the distal bowel (arrow).
Based on the “double-bubble” sign and CDO diagnosis, emergency laparotomy was performed 72 h post-birth. Preoperative nasogastric decompression yielded 50 ml of bilious fluid, substantially reducing abdominal distension. Intraoperative exploration confirmed membranous duodenal atresia (Figure 2A), and a duodenoduodenostomy was performed. During the procedure, a systematic exploration of the entire gastrointestinal tract (from the stomach to the rectum) was conducted, and no other obstructive lesions were identified. Incidentally, a 10.0 × 6.0 mm mass adherent to the boundary between the jejunum and its mesentery was identified (Figure 2B). Given potential complication risks, the mass was surgically resected. To ensure complete excision of the lesion and prevent residual pancreatic tissue, approximately 2 cm of the adjacent normal jejunal wall was resected along with the mass. Subsequently, primary jejunojejunostomy was performed using an end-to-end anastomosis technique to restore intestinal continuity. Histopathological examination of the resected specimen confirmed the diagnosis of MHP. Hematoxylin-eosin (H&E) staining of the HP tissue revealed typical acinar, ductal, and islet structures, consistent with Heinrich Type I (Figure 3).
Intraoperative findings. (A) Surgery confirmed duodenal obstruction caused by a membrane (arrow). (B) A photograph of the HP mass in situ shows a 10.0 mm × 6.0 mm yellowish soft tissue mass located at the boundary between the jejunum and mesentery, adhering to the serosal surface of the jejunum (arrow).
Histopathological analysis of the resected specimen. (A) Microscopically, the lesion consists of HP tissue (black arrow), including acini, islet cells, and HP tissue extending to the jejunal serosa (yellow arrow) (magnification × 200). (B) HP tissue and pancreatic ducts (black arrow) (magnification × 100). The histopathological features of the lesion are consistent with Heinrich Type I.
Postoperative laboratory investigations on day 3 revealed the following results: WBC 13.8 × 10⁹/L (45% neutrophils, 40% lymphocytes); RBC 4.9 × 10^12^/L; Hb 160.0 g/L; PLT 220.0× 10⁹/L; Na^+^ 136.0 mmol/L, K^+^ 3.9 mmol/L, Cl^−^ 96.0 mmol/L. Serum amylase was 65.0 U/L and lipase was 48.0 U/L. Hepatic and renal function panels were within normal limits. The postoperative recovery was uneventful. Prophylactic antibiotics were administered preoperatively and for the first three days after surgery. Minimal enteral feeding was initiated on postoperative day 3 and advanced to full feeding by day 7. The patient was discharged on postoperative day 8. Twelve-month follow-up ultrasound showed no evidence of recurrent HP. Written informed consent for the publication of this case report and any accompanying images was obtained from the neonate's parents prior to manuscript submission.
Discussion
3
HP refers to pancreatic parenchyma anatomically separate from the orthotopic pancreas, lacking continuity with the normal pancreas. First described by Jean Schultz in 1727, HP was not histologically confirmed until Klob's detailed characterization in 1859 (5). HP is typically discovered incidentally during autopsy, surgery, or endoscopy (1, 4). Current literature—primarily case reports and small series—lacks large clinicopathologic studies (6). This gap in the literature, combined with its frequent subclinical presentation, makes it difficult to determine the true incidence.
HP is more common in males than females, with peak onset typically occurring between ages 40 and 60 (3, 4). The most frequent sites for HP anomalies are the duodenum (30%), stomach (25%), and jejunum (15%), followed by Meckel's diverticulum (6%). Ileal involvement is rare (3%) (1). Dolan et al. (3) reported that among 212 patients with HP, 136 were male and 76 were female. The heterotopic tissue was located in the stomach (81 cases), duodenum (77 cases), or upper jejunum (33 cases). The remaining cases involved Meckel's diverticulum (6%), ileum (3%), and various other sites in small proportions. In contrast, Zhang et al. (4) observed a slight female predominance in a cohort of 184 patients with HP (male-to-female ratio: 0.94). This study found the stomach to be the most common location (97 cases, 52.7%), followed by the small intestine (48 cases, 26%), lesser omentum (18 cases, 10%), spleen/portal vein region (5 cases, 2.7%), mesentery (7 cases, 3.8%), distal esophagus (4 cases, 2.2%), hilar region (2 cases, 1%), and gallbladder (1 case, 0.5%). CDO-HP co-occurrence remains exceptionally rare, with pediatric MHP being particularly scarce—rarely reported in children <10 years and exceedingly rare in neonates (7).
Since most occurrences of HP are in the upper gastrointestinal tract, extraintestinal incidence is substantially lower. We searched PubMed from January 1980 to August 2025 using [(“heterotopic pancrea*” OR “ectopic pancrea*”) AND mesent*]. Reference lists of eligible articles were screened. We included English-language case reports/series describing mesenteric HP with clinical details; our case was also included. A total of 32 reports were identified (7–31) (Table 1). From the table, MHP demonstrated a striking female predominance (68.75% [22/32] female vs. 31.25% [10/32] male), contrasting sharply with the established male predominance in general HP. Anatomically, 78.125% (25/32) involved jejunal mesentery, while 12.5% (4/32) affected colonic mesentery. Three additional cases described MHP in small intestinal mesentery without precise localization (12, 15, 18). Pathologically, most were Heinrich type I, comprising acini, ducts, and islet cells.
CT enhancement patterns reflect HP's microscopic composition: acinar-dominant lesions show homogeneous enhancement similar to or exceeding normal pancreas, while ductal-dominant lesions exhibit heterogeneous, reduced enhancement (32, 33). MHP demonstrates imaging characteristics analogous to orthotopic pancreas. Jejunal MHP typically appears as an elongated, lobulated lesion with a broad jejunal base (34). Mesenteric lesions show higher long-to-short diameter ratios (3.0) than gastric/proximal small bowel lesions (1.4–1.5) (20). MHP maximum diameters exceed gastric HP (4.4 cm vs. 1.8–2.7 cm), with longitudinal jejunal alignment more frequent than at other HP sites (20). Histopathologically, microscopic HP infiltrates jejunal walls into muscularis propria (13). In our case, HP extended to the serosa (Figure 3), suggesting MHP may exhibit extra-tubular growth.
In our review of the literature on MHP, CECT and MRCP proved most diagnostically valuable. Most patients exhibited normal orthotopic pancreas, while the HP typically manifested acute pancreatitis symptoms (12, 14, 23, 27, 30). Additionally, two cases of HP-related malignant transformation have also been reported (10, 15). Nevertheless, even in these presentations, differentiation from alternative conditions remains essential. Key differential diagnoses include acute appendicitis, perforated appendicitis, Meckel's diverticulitis (8, 28, 31), and para-duodenal hernia (19). When evaluating suspected gastrointestinal mesenchymal tumors, carcinoid tumors, lymphomas, and metastases—which may similarly present as homogeneous, well-enhanced soft tissue masses within the mesentery—CECT demonstrates limited reliability for distinguishing MHP (29). Although this patient's preoperative serum amylase and lipase levels were within normal limits and the mesenteric heterotopic pancreas (MHP) showed no secondary organic lesions, the future risk of severe complications—such as pancreatitis or malignant transformation—cannot be excluded. Given this potential for serious disease and malignant progression, local resection of the MHP represents the optimal clinical strategy for long-term prognosis (9, 13, 15, 29).
It is noteworthy that congenital duodenal atresia is frequently associated with other congenital anomalies, with congenital heart disease and Down syndrome being the most common (35). Other associated malformations include renal abnormalities and biliary atresia (36, 37). Therefore, conducting relevant baseline screening is crucial for patients with congenital duodenal atresia, both preoperatively and postoperatively (37). We also noted that Song et al. (38) previously reported a case of congenital duodenal atresia with coexisting heterotopic pancreas (HP), mirroring the dual pathological findings observed in our patient. This similarity raises a fundamental embryological question: Could aberrant migration or differentiation of the pancreatic bud disrupt duodenal recanalization during embryonic development? Importantly, this hypothesis does not amount to an inference of a causal relationship; it merely represents a potential embryological mechanism (38).
Bilious vomiting accompanied by delayed meconium passage in a neonate represents a clinical emergency, necessitating immediate referral to a specialized neonatal surgical center to prevent life-threatening complications such as intestinal ischemia, perforation, or septic shock. In the evaluation of a neonatal mesenteric mass or cyst, key differential diagnoses should include congenital enteric cyst, mesenteric lymphangioma, simple cyst, neoplastic cyst, and infectious cyst. In the present case, the mass was histopathologically confirmed to be HP without signs of metaplasia. An open surgical approach was adopted for this case, rather than a laparoscopic procedure. This decision was made after thorough team discussion, considering our limited experience with neonatal laparoscopic surgery. The primary goals were to ensure the safety of the anastomosis, minimize the risk of intraoperative complications, and optimize postoperative outcomes. The procedure was performed after obtaining informed consent from the child's guardians.
In conclusion, given the potential association between HP and duodenal atresia in asymptomatic cases, we propose systematic screening for HP in all neonates with this condition. If identified, concurrent prophylactic resection of the HP lesion during the repair of duodenal atresia is recommended to mitigate the risk of long-term complications, including pancreatitis and gastrointestinal bleeding.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pearson S. Aberrant pancreas. Review of the literature and report of three cases, one of which produced common and pancreatic duct obstruction. Arch Surg. (1951) 63(2):168–86. 10.1001/ARCHSURG.1951.0125004017200614846476 · doi ↗ · pubmed ↗
- 2Alqahtani A Aljohani E Almadi F Billa S Alqahtani M Alkhaldi H. Heterotopic pancreatic tissue in the gastric antrum an incidental finding during bariatric surgery: a case report and literature review. Int J Surg Case Rep. (2020) 67:39–41. 10.1016/j.ijscr.2019.12.04032004902 PMC 7076269 · doi ↗ · pubmed ↗
- 3Dolan RV Re Mine WH Dockerty MB. The fate of heterotopic pancreatic tissue. A study of 212 cases. Arch Surg. (1974) 109(6):762–5. 10.1001/archsurg.1974.013600600320104420439 · doi ↗ · pubmed ↗
- 4Zhang Y Sun X Gold JS Sun Q Lv Y Li Q Heterotopic pancreas: a clinicopathological study of 184 cases from a single high-volume medical center in China. Hum Pathol. (2016) 55:135–42. 10.1016/j.humpath.2016.05.00427195908 · doi ↗ · pubmed ↗
- 5Le Compte MT Mason B Robbins KJ Yano M Chatterjee D Fields RC Clinical classification of symptomatic heterotopic pancreas of the stomach and duodenum: a case series and systematic literature review. World J Gastroenterol. (2022) 28(14):1455–78. 10.3748/wjg.v 28.i 14.145535582670 PMC 9048474 · doi ↗ · pubmed ↗
- 6Zhang X Peng L Wang Z Pan F Ren R Li Y Extensive heterotopic pancreas in a rare site: a case report and a review of literature. Medicine (Baltimore). (2023) 102(9):e 32241. 10.1097/MD.000000000003224136862885 PMC 9981405 · doi ↗ · pubmed ↗
- 7Koltai IL Hoffmann-von Kap-herr S Weitzel D. [Accessory pancreatic cyst located in the mesocolon in a child (author’s transl)]. Z Kinderchir. (1981) 32(3):277–82. 10.1055/s-2008-10632707282061 · doi ↗ · pubmed ↗
- 8Fam SO’Briain DS Borger JA. Ectopic pancreas with acute inflammation. J Pediatr Surg. (1982) 17(1):86–7. 10.1016/s 0022-3468(82)80338-27077488 · doi ↗ · pubmed ↗