PseudotimeDE-fast: fast testing of differential gene expression along cell pseudotime
Yuheng Lai, Dongyuan Song, Lucy Xia, Jingyi Jessica Li

TL;DR
PseudotimeDE-fast is a new method for quickly and accurately finding genes that change expression over time in single-cell RNA sequencing data.
Contribution
PseudotimeDE-fast introduces a scalable and computationally efficient approach for differential gene expression analysis along pseudotime with well-calibrated P-values.
Findings
PseudotimeDE-fast delivers comparable or better performance than existing methods.
The method offers substantial improvements in computational efficiency.
It handles uncertainty in pseudotime inference to produce valid P-values.
Abstract
Identifying differentially expressed (DE) genes along cell pseudotime is crucial for understanding dynamic biological processes captured by single-cell RNA sequencing. However, existing DE methods either produce invalid P-values by ignoring the uncertainty in pseudotime inference or struggle to scale with the growing size of modern datasets. To address these limitations, we introduce PseudotimeDE-fast, a scalable method for detecting DE genes along pseudotime with well-calibrated P-values. Through comprehensive simulations and real-data analyses, we demonstrate that PseudotimeDE-fast delivers comparable or superior performance to existing approaches while offering substantial improvements in computational efficiency. PseudotimeDE-fast is implemented in R with Rcpp acceleration and released under the MIT license. The source code is available at: https://github.com/dsong-lab/PseudotimeDE.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
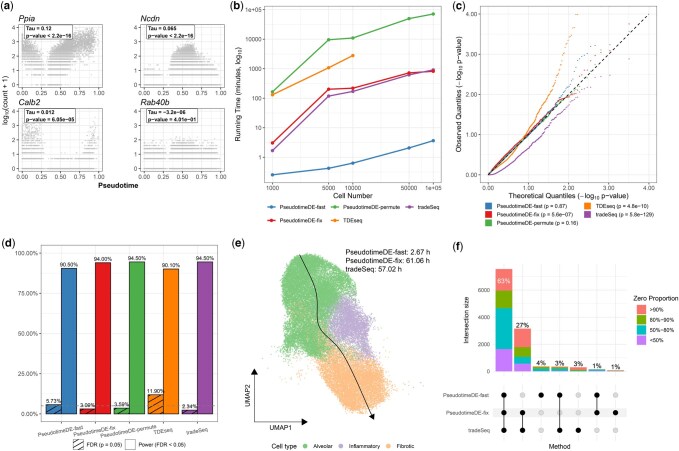 Figure 1
Figure 1- —National Science Foundation10.13039/100000001
- —Chan Zuckerberg Initiative Donor Advised Fund
- —Silicon Valley Community Foundation10.13039/100000923
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSingle-cell and spatial transcriptomics · Gene expression and cancer classification · Gene Regulatory Network Analysis
1 Introduction
Single-cell RNA sequencing (scRNA-seq) technologies have become a powerful tool for uncovering continuous transitions in cell populations. A common approach involves inferring a latent temporal variable, known as “pseudotime,” from gene expression profiles to represent cells’ relative positions along a developmental trajectory (Trapnell et al. 2014). To interpret pseudotime, differential expression (DE) analysis is typically performed to identify genes with significant expression changes along the trajectory. Several methods have been developed for this purpose, such as tradeSeq (Van den Berge et al. 2020), scMaSigPro (Srivastava et al. 2024), and TDEseq (Fan et al. 2024). However, these methods rely on regression models that treat pseudotime as fixed, ignoring the uncertainty in its inference. This oversight can lead to invalid P-values, as shown in prior studies (Campbell and Yau 2016, Song and Li 2021).
To consider the uncertainty in inferred pseudotime, we previously developed PseudotimeDE (Song and Li 2021), the first DE method to explicitly account for this uncertainty. PseudotimeDE repeatedly performs trajectory (pseudotime) inference on subsampled cells and applies permutations to break the gene expression–pseudotime association, fitting a regression model to generate a null distribution of the test statistic. This approach yields well-calibrated P-values and good statistical power. However, its extensive computational demands, due to repeated model fitting on many subsamples, limit its scalability and broader adoption in the single-cell community.
To overcome the computational limitations of PseudotimeDE, we propose PseudotimeDE-fast, a novel method and updated R package for fast testing of gene expression changes along cell pseudotime. Unlike the methods that rely on regression models assuming fixed pseudotime, PseudotimeDE-fast tests the independence between gene expression and pseudotime by treating both as random variables. It implements a hypothesis test using a novel adaptation of the Bergsma–Dassios sign covariance —a robust extension of Kendall’s tau—for sparse data, where if and only if the two variables are independent (Bergsma and Dassios 2014). Through comprehensive simulations and analysis of a large real dataset, we show that PseudotimeDE-fast produces well-calibrated P-values, achieves comparable or improved FDR control and power, and is over 100 times faster than existing methods.
2 Implementation
PseudotimeDE-fast is implemented in R and can be installed via devtools::install_github(“dsong-lab/PseudotimeDE”). To address the computational bottleneck of its predecessor PseudotimeDE, it replaces the subsampling-and-permutation procedure with a direct, deterministic statistical test. Specifically, it reframes DE analysis as a formal test of independence between the pseudotime vector X and the expression vector of gene g.
The input consists of a scRNA-seq count matrix , where n is the number of cells and p is the number of genes, and a pseudotime vector representing the inferred pseudotime of cells. For each gene , PseudotimeDE-fast efficiently computes , a consistent estimator of the Bergsma–Dassios sign covariance :
where
The intuition for this measure, a powerful extension of the well-known Kendall’s (Kendall 1938), is that it moves beyond comparing simple pairs of points to evaluating all sets of four points (quartets). For each quartet, it checks whether the arrangement of points is “concordant” or “discordant” for both pseudotime and gene expression.
Previously, Heller and Heller (2016) introduced an algorithm to compute with complexity, which becomes computationally prohibitive as n (the number of cells) increases. To address this, we developed an optimized algorithm that reduces the complexity of its core step to , where denotes the number of unique expression levels, often small due to sparsity in scRNA-seq data. Details are provided in Supplementary Material 1, available as supplementary data at Bioinformatics online. Under the null hypothesis of independence between X and , admits a known limiting distribution, enabling efficient hypothesis testing (Nandy et al. 2016). Compared to other rank-based independence tests with similar statistical properties (Shi et al. 2022), our implementation achieves near-linear scalability for sparse data, while existing methods typically face computational bottlenecks.
3 Results
To evaluate the performance of PseudotimeDE-fast in terms of runtime, P-value validity, FDR control, and statistical power for detecting DE genes, we conducted simulations across varying numbers of cells (n) and used a large-scale real scRNA-seq dataset (Tsukui et al. 2024). We compared PseudotimeDE-fast with state-of-the-art trajectory-based DE methods, including PseudotimeDE (Song and Li 2021)—in both its asymptotic (fix) mode, which ignores pseudotime uncertainty and is not recommended, and its subsampling-and-permutation (permute) mode, which is accurate but computationally intensive—as well as tradeSeq (Van den Berge et al. 2020) and TDEseq (Fan et al. 2024). The details about the implementation and computational resources are described in Supplementary Material 2, available as supplementary data at Bioinformatics online.
We generated synthetic datasets with p = 2000 genes (20% DE) and varying numbers of cells n ∈ {1000, 5000, 10 000, 50 000, 100 000} using scDesign3 (Song et al. 2024), which was trained on a real scRNA-seq dataset of dentate gyrus neurogenesis (Hochgerner et al. 2018). Figure 1a shows results for four example genes: PseudotimeDE-fast reported highly significant P-values for three DE genes (Ppia, Ncdn, and Calb2) and an insignificant P-value for a non-DE gene (Rab40b).
Benchmarking PseudotimeDE-fast against other trajectory-based DE methods. (a) Expression of four example genes along pseudotime. The estimated τn and corresponding P-values are shown for each gene. (b) Runtime comparison across different cell numbers (n). PseudotimeDE-fast is significantly faster than all other methods. (c) Quantile-quantile plots of P-values under the null hypothesis on the − log 10 scale. Only PseudotimeDE-fast and PseudotimeDE-permute produce well-calibrated P-values, with points falling along the diagonal and Kolmogorov-Smirnov test P>.05. (d) FDR and power in simulations. PseudotimeDE-fast achieves reasonable FDR control and comparable power to existing methods. (e) Application to the alveolar fibroblast lineage dataset (Tsukui et al. 2024). Cells are visualized by UMAP; colors denote cell types, and the curve indicates the inferred trajectory. PseudotimeDE-fast is 30× faster than PseudotimeDE-fix and tradeSeq. (f) UpSet plot showing overlaps in identified DE genes. PseudotimeDE-fast shares 63% of DE genes with both other methods, indicating high consistency.*
Figure 1b compares runtime across methods as n increases. All methods support multi-core parallelization, so we set the number of CPUs as 10 for every method. At n = 10 000, PseudotimeDE-fast completed in 124.29 s (CPU time): 298 times faster than tradeSeq, 348 times faster than PseudotimeDE-fix, 4408 times faster than TDEseq, and over 24 013 times faster than PseudotimeDE-permute. In terms of clock time, PseudotimeDE-fast finished in just 26.8 s. Note that TDEseq failed to finish within a reasonable runtime (48 h) with n=50 000 or more cells (Supplementary Material 2, available as supplementary data at Bioinformatics online).
To assess P-value validity under the null, we compared P-values to the distribution in two ways: (i) quantile-quantile (QQ) plots using P-values, and (ii) Kolmogorov–Smirnov tests using the raw P-values (Fig. 1c). PseudotimeDE-fast and PseudotimeDE-permute yielded well-calibrated P-values close to the expected uniform distribution. For DE gene detection at n=10 000 (additional results in Fig. 1, available as supplementary data at Bioinformatics online), PseudotimeDE-fast achieved comparable power and FDR control to state-of-the-art methods while using far less computational time (Fig. 1d). For PseudotimeDE-fix, although its FDR was controlled, its P-values showed deviation from the expected uniform distribution (Fig. 1c; Fig. 2, available as supplementary data at Bioinformatics online). In addition, although PseudotimeDE-fast showed a slight power loss compared to PseudotimeDE-permute, the few missed genes were highly sparse and often of limited biological interest (Fig. 3, available as supplementary data at Bioinformatics online). These results highlight PseudotimeDE-fast as a scalable solution for large-scale pseudotime DE analysis. Note that this simulation has a high signal-to-noise ratio, so pseudotime can be estimated accurately and the “double-dipping” issue (Neufeld et al. 2024) is relatively mild. If double-dipping remains a major concern, PseudotimeDE-fast may be combined with the synthetic-null-data approach used by ClusterDE (Song et al. 2025) to improve FDR control.
We further evaluated PseudotimeDE-fast using a large-scale scRNA-seq dataset of alveolar fibroblast lineage comprising n= 35 096 cells and p=12 834 genes (Tsukui et al. 2024). This dataset contains a single trajectory, and pseudotime was inferred using Slingshot (Street et al. 2018). We applied PseudotimeDE-fast, PseudotimeDE-fix, and tradeSeq, which are the only feasible methods for this dataset, and excluded PseudotimeDE-permute and TDEseq due to scalability issues. PseudotimeDE-fast completed the analysis in under 3 h, making it over 30 times faster than the other two methods, each of which required more than two days (Fig. 1e).
Since ground-truth DE genes are unknown, we assessed consistency across methods as a proxy for power. PseudotimeDE-fast identified a largely overlapping set of DE genes, sharing 63% with both other methods (Fig. 1f). Among DE genes missed by PseudotimeDE-fast but detected by both other methods (27%), 66.1% had zero expression in over 80% of cells, indicating high sparsity and limited informativeness. These results highlight that PseudotimeDE-fast offers substantial speed gains while maintaining good statistical power to existing approaches.
4 Discussion
Based on the Bergsma–Dassios sign covariance (an association measure for two random variables), PseudotimeDE-fast does not natively adjust for covariates such as batch effects or sequencing depth; users should therefore correct for confounders prior to analysis. Extending PseudotimeDE-fast to handle covariates would require a conditional (or partial) form of the Bergsma–Dassios sign covariance, which, to our knowledge, has not yet been developed and represents an interesting direction for future research.
Supplementary Material
btaf573_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bergsma W , Dassios A. A consistent test of independence based on a sign covariance related to Kendall’s tau. Bernoulli 2014;20:1006–28.
- 2Campbell KR , Yau C. Order under uncertainty: robust differential expression analysis using probabilistic models for pseudotime inference. P Lo S Comput Biol 2016;12:e 1005212.27870852 10.1371/journal.pcbi.1005212 PMC 5117567 · doi ↗ · pubmed ↗
- 3Fan Y , Li L, Sun S et al Powerful and accurate detection of temporal gene expression patterns from multi-sample multi-stage single-cell transcriptomics data with TD Eseq. Genome Biol 2024;25:96.38622747 10.1186/s 13059-024-03237-3PMC 11020788 · doi ↗ · pubmed ↗
- 4Heller Y , Heller R. Computing the bergsma dassios sign-covariance. ar Xiv, ar Xiv:1605.08732, 2016, preprint: not peer reviewed.
- 5Hochgerner H , Zeisel A, Lönnerberg P et al Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat Neurosci 2018;21:290–9.29335606 10.1038/s 41593-017-0056-2 · doi ↗ · pubmed ↗
- 6Kendall MG. A new measure of rank correlation. Biometrika 1938;30:81–93.
- 7Nandy P , Weihs L, Drton M et al Large-sample theory for the Bergsma-Dassios sign covariance. Electron J Statist 2016;10:2287–311.
- 8Neufeld A , Gao LL, Popp J et al Inference after latent variable estimation for single-cell RNA sequencing data. Biostatistics 2024;25:270–87.10.1093/biostatistics/kxac 047PMC 1223526036511385 · doi ↗ · pubmed ↗