Near-complete genome of two genotype II African swine fever viruses recovered from domestic pigs in Tanzania
Jean N. Hakizimana, Clara Yona, Charles Kayuki, Mariam R. Makange, Ester K. Adamson, Amanda Warr, Christine Tait-Burkard, Hans J. Nauwynck, Gerald Misinzo

TL;DR
This study sequenced two nearly complete genomes of a specific type of African swine fever virus found in pigs in Tanzania.
Contribution
The study provides new genomic data of genotype II African swine fever viruses from Tanzania using nanopore sequencing.
Findings
Two near-complete genomes of genotype II ASFV were sequenced from domestic pigs in Tanzania.
The genomes were closely related to other genotype II isolates globally.
Abstract
Two near-complete genomes of genotype II African swine fever viruses (ASFV) recovered from domestic pigs in Mbozi district, Tanzania, in 2017 were generated using tiled amplicon Oxford nanopore sequencing. These two ASFV genomes described in this study were closely related to other genotype II isolates reported worldwide.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
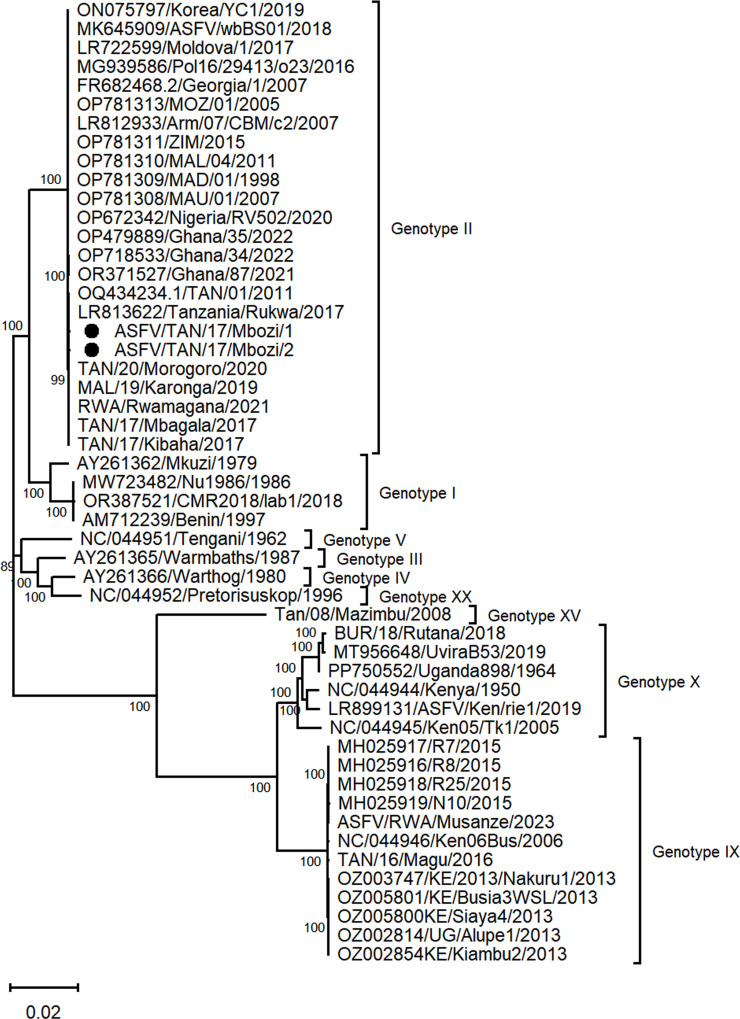 Fig 1
Fig 1| Isolate ID | Total | ASFV | N50 | Percentage of ASFV specific reads (%) | Mean | Assembled | Mean | GC content (%) | Accession number |
|---|---|---|---|---|---|---|---|---|---|
| ASFV/TAN/17/Mbozi/1 | 245,015 | 212,621 | 6,930 | 86.78 | 11.5 | 172,585 | 2,500 | 38.95 |
|
| ASFV/TAN/17/Mbozi/2 | 482,432 | 419,948 | 6,031 | 87.056 | 11.7 | 167,022 | 4,370 | 39.17 |
|
- —RSIF_JIRA-PASET
- —Oliver R. Tambo Africa Research Chairs Initiative
- —International Foundation for Sciencehttp://dx.doi.org/10.13039/100004413
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Disease Management and Epidemiology · Vector-Borne Animal Diseases · Viral Infections and Immunology Research
ANNOUNCEMENT
African swine fever (ASF) is a transboundary animal disease with a mortality rate reaching 100% (1). Whole genome sequencing of ASF virus (ASFV) provides insights for the design of control strategies. The ASFV is an enveloped, double-stranded DNA virus belonging to the family Asfarviridae, genus Asfivirus (2). To overcome ASFV sequencing challenges, including the dominance of host DNA, a tiled amplicon sequencing approach has been developed (3, 4).
Tissue samples, including spleen and mesenteric lymph nodes, collected during the 2017 ASF outbreak in Mbozi district in southern Tanzania (ASFV/TAN/17/Mbozi/1 and ASFV/TAN/17/Mbozi/2) were tested for ASFV and clustered within ASFV genotype II as previously described (5). The DNA was extracted using the QIAamp DNA purification kit (Qiagen, Hilden, Germany), followed by polymerase chain reaction (PCR) amplification as previously described (3). A total of 32 tiled primer pairs were used to amplify fragments of about 7 kb with overlaps of 1 kb between adjacent amplicons using PCR Bio VeriFi Hot Start high-fidelity DNA polymerase. Amplicon size was verified by 1% agarose gel electrophoresis (Gel DocTM EZ Imager, Bio-Rad, Hercules, CA). The amplicons were pooled for library preparation using the ligation sequencing kit (SQK-LSK109, Oxford Nanopore Technologies, Oxford, UK) and sequenced on an R9.4.1 flow cell using MinION MK1c with basecalling and demultiplexing performed by Guppy v5.0.14 (Oxford Nanopore Technologies, Oxford, UK). The Lilo pipeline (https://github.com/amandawarr/Lilo) was used for data analysis. Lilo uses a reference to sort the amplicons and separate reads into amplicons by alignment position using bedtools v2.30.0, followed by polishing against the highest quality reads, while primer sequences are removed using Porechop v0.2.3, and assembly is performed with Scaffold_builder v2.3. In this study, the Lilo pipeline was modified due to some misassembly caused by chimeric amplicons in the sequencing data. In the rule “assign”, bedtools intersect command parameters were edited to include “-F 0.85 f 0.85” to limit the selection to sequences that contained only the target amplicon, while allowing flexibility for real indels. The resulting assemblies were evaluated using the Quality Assessment Tool (QUAST) version 5.0.2 (6).
The nucleotide sequences of the assembled ASFV strains had a genome size of 172,585 and 167,022 base pairs (bp), average coverage of 2,500 and 4,370 reads per nucleotide, and a GC content of 38.95 and 39.17% for the strains ASFV/TAN/17/Mbozi/1 and ASFV/TAN/17/Mbozi/2, respectively (Table 1). After the NCBI GenBank database search and phylogenetic reconstruction (Fig. 1), the sequences described in this study were closely related to the TAN/01/2011 ASFV isolate (OQ434234) (7) belonging to genotype II with a nucleotide identity of 99.82% and a query coverage of 100%. In addition to the lack of the highly repetitive 3′- and 5′ telomeric regions for the two sequences, indels were observed along the alignment after comparison with the ASFV genotype II reference genome Georgia2007/1 (FR682468.2), including a deletion of fragments of 5,465 and 5,220 bp at positions 16,190–21,665 and 169,120–174,340, respectively, leading to the truncation of the MGF 360-1Lb CDS and ASFV G ACD 01980 CDS in both ASFV strains described in this study.
Maximum Likelihood phylogenetic tree reconstructed using ASFV complete genome nucleotide sequences including those described in this study (marked by a black circle) and those previously reported worldwide available at the NCBI GenBank. The phylogeny was inferred using Tamura (992) nucleotide substitution model, as determined by the Bayesian Information Criterion model selection analysis. The sequences were aligned using MAFFT version 7.221 and phylogenetic tree reconstructed using 1,000 bootstrap replications as implemented in MEGA 12. The scale bar indicates nucleotide substitution per site and the node values show the percentage of bootstrap support.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pikalo J, Zani L, Hühr J, Beer M, Blome S. 2019. Pathogenesis of African swine fever in domestic pigs and European wild boar - Lessons learned from recent animal trials. Virus Res 271:197614. doi:10.1016/j.virusres.2019.04.00130953662 · doi ↗ · pubmed ↗
- 2Alonso C, Borca M, Dixon L, Revilla Y, Rodriguez F, Escribano JM, Ictv Report Consortium. 2018. ICTV virus taxonomy profile: Asfarviridae. J Gen Virol 99:613–614. doi:10.1099/jgv.0.00104929565243 PMC 12662184 · doi ↗ · pubmed ↗
- 3Warr A, Newman C, Craig N, Vendelė I, Pilare R, Cruz LC, Barangan TG, Morales RG, Opriessnig T, Venturina VM, Mananggit MR, Lycett S, Domingo CY, Tait-Burkard C. 2021. No part gets left behind: tiled nanopore sequencing of whole ASFV genomes stitched together using Lilo. bio Rxiv. doi:10.1101/2021.12.01.470769 · doi ↗
- 4Oxford Nanopore Technologies. 2022. PCR Tiling of African Swine Fever (ASF) Virus (SQK-LSK 109 with EXP-NBD 104 or EXP-NBD 114). Oxford Nanopore Technologies. Available from: https://nanoporetech.com/document/pcr-tiling-of-african-swine-fever-virus
- 5Yona CM, Vanhee M, Simulundu E, Makange M, Nauwynck HJ, Misinzo G. 2020. Persistent domestic circulation of African swine fever virus in Tanzania, 2015-2017. BMC Vet Res 16:369. doi:10.1186/s 12917-020-02588-w 33004025 PMC 7528248 · doi ↗ · pubmed ↗
- 6Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi:10.1093/bioinformatics/btt 08623422339 PMC 3624806 · doi ↗ · pubmed ↗
- 7Mthombeni RF, Bastos AD, van Schalkwyk A, van Emmenes J, Heath L. 2023. Phylogenomic comparison of seven African swine fever genotype II outbreak viruses (1998-2019) reveals the likely African origin of Georgia 2007/1. Pathogens 12:1129. doi:10.3390/pathogens 1209112937764936 PMC 10537866 · doi ↗ · pubmed ↗