High-quality draft genome sequence of the tropical lichen-forming fungus Lecanora helva (Lecanoraceae, Ascomycota)
Yukun Sun, Meredith M. Doellman, Alejandrina Barcenas-Peña, Vasun Poengsungnoen, Sabine Huhndorf, H. Thorsten Lumbsch, Felix Grewe

TL;DR
This paper provides a high-quality genome sequence of the lichen-forming fungus Lecanora helva from a tropical mangrove in Thailand.
Contribution
The study introduces a novel multi-assembler approach to generate a nearly complete genome sequence of Lecanora helva.
Findings
The genome assembly is 30.07 Mb with 98.1% BUSCO completeness.
The sequencing was done using PacBio HiFi technology.
The genome will support research on mangrove adaptation and climate resilience.
Abstract
We present a high-quality genome assembly of the lichenized fungus Lecanora helva collected in a tropical mangrove in Thailand. Using PacBio HiFi sequencing and a novel multi-assembler approach, we generated a contiguous, nearly complete (30.07 Mb, 98.1% BUSCO completeness) genome sequence, enabling future studies on mangrove adaptation and climate resilience.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
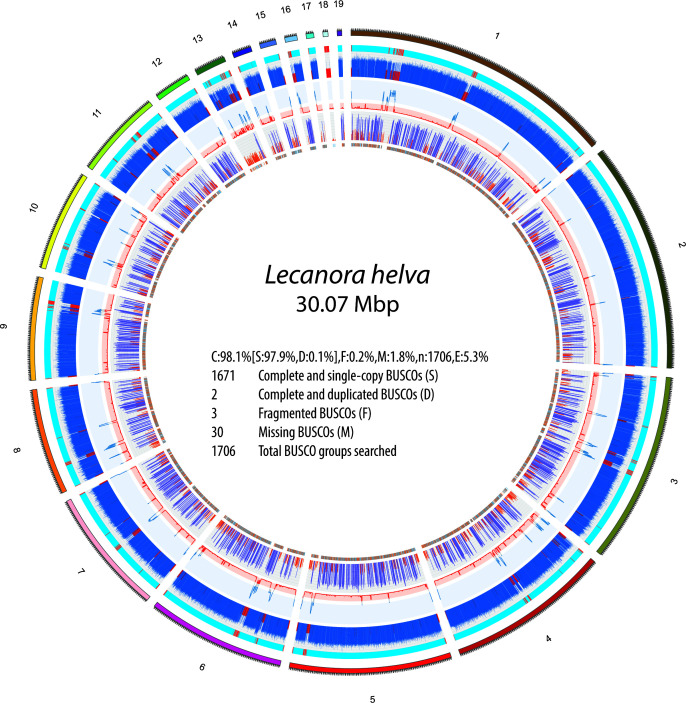 Fig 1
Fig 1- —Grainger Foundationhttp://dx.doi.org/10.13039/100008074
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Microbial Community Ecology and Physiology · Plant Pathogens and Fungal Diseases
ANNOUNCEMENT
Lecanora is one of the largest genera of crustose lichens, with a worldwide distribution (1, 2). We sequenced the genome of the fungal component (mycobiont) of Lecanora helva Stizenb., a tropical lichen species potentially sensitive to climate change. The specimen was collected on 07 August 2021 from the bark of Avicennia alba Blume in a mangrove forest in Tambon Lat Khwang, Amphoe Ban Pho, Chachoengsao Province, Thailand (13^o^36’41.9”N, 101^o^02’21.4”E), and deposited in the Lichen Herbarium of Ramkhamhaeng University (accession: RAMK 036147; collection number: VPMG_37; herbarium database: http://www.lichen.ru.ac.th/images/data/STT/STT46_Lichen%20herbarium%20database%20and%20management.pdf). An axenic fungal culture was produced from a single ascospore and grown on malt-yeast extract agar under laboratory conditions (3).
High-molecular-weight DNA was extracted from the axenic fungal culture using a modified protocol in Wilken et al. (4) and sent to the UWBC DNA Sequencing Facility. DNA quality and quantity were measured using a NanoDrop One (ThermoFisher Scientific, Waltham, MA, USA), a Qubit dsDNA High Sensitivity kit, and a FemtoPulse System (Agilent, Santa Clara, CA, USA). A PacBio HiFi library was prepared using protocol PN 102-166-600 APR2022 (Pacific Biosciences, Menlo Park, CA, USA), with modifications including Covaris gTUBE shearing (Covaris, LLC, Woburn, MA, USA) without further size selection. Library quality was assessed using the FemtoPulse, and the library was quantified using the Qubit dsDNA High Sensitivity kit. Sequencing was performed on a Sequel II using the Sequel Polymerase Binding Kit 2.2. A total of 620,494 raw reads were generated (N_50_ =8148); adapter contamination (“AAGCAGTGGTATCAACGCAGAGTACT”) was removed with lima v.2.7.1, and duplicates were marked with pbmarkdup v.1.0.3 (5).
The assembly was constructed with a novel consensus-based strategy combining outputs from multiple assemblers. PacBio HiFi reads were assembled into unphased contigs using six tools: Canu v.2.2 (6), NextDenovo v.2.5.2 (7), IPA (8), Flye v.2.9.5-b1801 (9), Peregrine-2021 v.0.4.13 (10), and RAFT-hifiasm v.0.19.9-r616 (11–14). The Peregrine-2021 output was selected as the primary assembly due to its lowest contig number, highest N_50_, and greater number of identified telomeric ends, indicating superior genome contiguity. To obtain additional telomere-to-telomere contigs, contigs with telomeric sequences at one terminus from alternative assemblies were compared and merged with QuickMerge v.0.3 (15). Resulting contigs were manually curated; Redundans v2.0.1 (16) was used for redundancy reduction. Genome contiguity and completeness were assessed using QUAST v.5.3.0 and BUSCO v.5.7.0 with the ascomycota_odb10 data set (17, 18). Repeats were identified and masked before annotation using RepeatModeler 2.0.1 (19), followed by RepeatMasker 4.1.2-p1 to mask repetitive elements (20). Annotation was performed using Funannotate v.1.8.17 and InterProScan v.5.72-103.0 (21, 22). Default parameters were used except where otherwise noted.
Our final L. helva genome assembly is 30.07 Mb in total length, distributed across 19 contigs, with an N_50_ of 2.68 Mb (Fig. 1). The estimated coverage is 140.99 ×, and the GC content is 46.87%. BUSCO analysis indicated 98.1%. A total of 9,745 protein-coding genes were predicted. Our assembly method, which leverages the strengths of multiple assemblers, will support further work on the adaptation of L. helva to mangrove habitats and the conservation of tropical mangrove forest diversity in the face of climate change.
Circular representation of L. helva genomic features. The plot comprises concentric tracks, organized from inner to outer rings: gene locations, with forward strand genes in orange and reverse strand genes in blue; gene density (1/10 k base pair), highlighting regions with high density (>0.7) in purple and low density (<0.3) in orange; repeat density (1/10 k base pair), illustrating areas with high repeat content (>0.7) in blue and low repeat content (<0.3) in red; GC content, marking regions with high GC content (>0.5) in blue and low GC content (<0.3) in red; and GC regions, identifying AT-rich regions (with GC content 0–37.9%) in red and higher GC content (37.9–100%) in cyan. In the center of the plot, the BUSCO completeness assessment of the assembled genome using the Ascomycota lineage data set (ascomycota_odb10) is displayed. Scores indicate the percentages of complete (single-copy and duplicated), fragmented, and missing orthologs.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Papong K, Lumbsch HT. 2011. A taxonomic survey of Lecanora sensu stricto in Thailand (Lecanoraceae; Ascomycota). The Lichenologist 43:299–320. doi:10.1017/S 0024282911000247 · doi ↗
- 2Papong K, Boonpragob K, Parnmen S, Lumbsch HT. 2013. Molecular phylogenetic studies on tropical species of Lecanora sensu stricto (Lecanoraceae, Ascomycota). nova_hedwigia 96:1–13. doi:10.1127/0029-5035/2012/0072 · doi ↗
- 3Rosabal D, Pino-Bodas R. 2024. A review of laboratory requirements to culture lichen mycobiont species. J Fungi (Basel) 10:621. doi:10.3390/jof 1009062139330381 PMC 11433509 · doi ↗ · pubmed ↗
- 4Wilken PM, Aylward J, Chand R, Grewe F, Lane FA, Sinha S, Ametrano C, Distefano I, Divakar PK, Duong TA, Huhndorf S, Kharwar RN, Lumbsch HT, Navathe S, Pérez CA, Ramírez-Berrutti N, Sharma R, Sun Y, Wingfield BD, Wingfield MJ. 2020. IMA Genome - F 13. IMA Fungus 11:19. doi:10.1186/s 43008-020-00039-733014691 PMC 7513301 · doi ↗ · pubmed ↗
- 5Armintoepfer A. 2020. pbmarkdup: Mark duplicate reads from Pac Bio sequencing of an amplified library. Git Hub. Available from: https://github.com/Pacific Biosciences/pbmarkdup. Retrieved 20 Jul 2022.
- 6Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, Miga KH, Eichler EE, Phillippy AM, Koren S. 2020. Hi Canu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res 30:1291–1305. doi:10.1101/gr.263566.12032801147 PMC 7545148 · doi ↗ · pubmed ↗
- 7Hu J, Wang Z, Sun Z, Hu B, Ayoola AO, Liang F, Li J, Sandoval JR, Cooper DN, Ye K, Ruan J, Xiao C-L, Wang D, Wu D-D, Wang S. 2024. Next Denovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol 25:107. doi:10.1186/s 13059-024-03252-438671502 PMC 11046930 · doi ↗ · pubmed ↗
- 8Pacific Biosciences. 2022. pbipa: Improved Phased Assembler (v 1.8.0). Git Hub. Available from: https://github.com/Pacific Biosciences/pbipa. Retrieved 10 Jun 2024.