Genome sequence and methylome of the extremely halophilic bacterium Salinibacter ruber strain M31T isolated from a crystallizer pond in Mallorca, Spain
Beatriz Toledo Akiti, Gülay Kaya, Sean P. Kennedy, Priya DasSarma, Tamas Vincze, Alexey Fomenkov, Richard J. Roberts, Shiladitya DasSarma

TL;DR
This paper presents the genome and methylome of Salinibacter ruber, a salt-loving bacterium from Spain, revealing its unique genetic features.
Contribution
The study provides the first complete genome and methylome of Salinibacter ruber strain M31T, including insights into its archaeal-like proteins.
Findings
The genome consists of a 3.55-Mbp chromosome and a 35.5-kbp plasmid.
The proteome is highly acidic and includes archaeal-like proteins.
Single-molecule real-time sequencing was used to analyze the genome and methylome.
Abstract
Salinibacter ruber strain M31T, an extremely halophilic bacterium, was isolated from a saltern crystallizer pond in Spain. Single-molecule real-time sequencing revealed a 3.6-Mbp genome with a single 3.55-Mbp circular chromosome and a 35.5-kbp plasmid. The highly acidic proteome includes a total of 2,962 proteins, some of which are archaeal-like.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
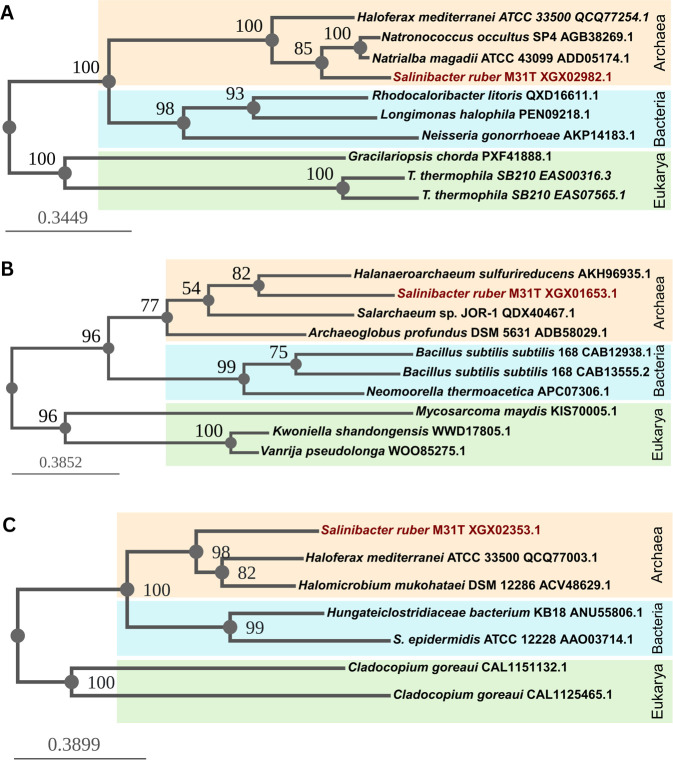 Fig 1
Fig 1| Replicon properties | Chromosome | pSR35 | Whole genome |
|---|---|---|---|
| Size (bp) | 3,551,259 | 35,506 | 3,586,765 |
| GC content (%) | 66.2 | 57.9 | 66.1 |
| Coverage | 104 | 147 | 104 |
| Gene no. | 3,015 | 29 | 3,044 |
| Encoded proteins no. | 2934 | 28 | 2962 |
| GenBank |
|
|
|
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Bacteriophages and microbial interactions · Microbial Community Ecology and Physiology
ANNOUNCEMENT
Salinibacter ruber is an extremely halophilic bacterium that represents an interesting model for convergent evolution with halophilic Archaea as well as astrobiological studies (1–3). Besides an elevated intracellular concentration of KCl for osmotic balance, it also possesses rhodopsins for phototrophic energy production and displays a high degree of genome plasticity (4). The gram-negative Salinibacter ruber strain M31^T^ was isolated from a saltern crystallizer pond in Mallorca, Balearic Islands, Spain (GPS: 39.3499° N, 3.0110° E) on September 1999 and was a gift from the American Type Culture Collection (1). Salinibacter ruber M31^T^ cultures were grown in ATCC 2402 medium (5). High-molecular weight genomic DNA was prepared by lysis and extraction with phenol:chloroform, followed by ethanol precipitation as described earlier (6).
DNA samples (~2 µg) were sheared to an average size of ~10 kb using the G-tube protocol (Covaris, MA). DNA libraries were prepared using a SMRTbell Express Template Prep Kit 2.0 (100-938-900, Pacific Biosciences, CA) and ligated with barcoded hairpin lbc--lbc adapters for multiplex sequencing on a Sequel II (SQ2) instrument. Incompletely formed SMRTbell templates were removed by digestion with a combination of exonucleases III and VII according to the manufacturer’s instructions (NEB, MA). The qualification and quantification of the SMRTbell libraries were made on a Qubit fluorometer (Invitrogen, OR) and a 2100 bioanalyzer (Agilent Technologies, CA). Size-selected separation of SMRTbell libraries on the gel-based BluePippin instrument (Sage Science, Beverly, MA) was performed to remove impurities and improve genome assembly results. Single-molecule real-time (SMRT) sequencing was performed on a PacBio Sequel II platform (Pacific Biosciences, CA) with a 2 h pre-extension and 30 h collection time. Sequencing reads were assembled de novo using a microbial genome annotation analysis pipeline (SMRT Link v11.1.0.166339) with default parameters. The 60,343 HiFi (CCS) reads with a 6,165 bp mean read length and 6,767-bp N50 read length yielded 374 Mb of high-quality sequencing data. The polished assembly resolved automatically into a large circular chromosome and circular plasmid pSR35 (Table 1).
Genome annotation was performed using GeneMark.hmm2, Prokka, and National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline build 3190 (7–9). The mean isoelectric point (pI) of 5.61 calculated using IPC 2.0 indicates an acidic proteome which, together with the GC-rich genome (Table 1), is characteristic of extreme halophiles (10–12). The genome includes a single rRNA operon and 44 tRNA genes predicted using tRNAscan-SE 2.0 (13). Based on analysis using BLAST, NCBI’s annotation and HaloWeb, two archaeal-like sensory rhodopsins I, a halorhodopsin, a xanthorhodopsin, four photolyases, as well as 50 annotated transposases—only one in pSR35—are located in its genome (14–16). Consistent with adaptation to their evaporitic environment, stress and heavy metal proteins are also predicted. Additionally, phylogenetic analysis using BLAST and Phylogeny.fr’s advanced workflow identified possible candidates for archaeal lateral transfer origin (Fig. 1) (14, 17).
Maximum likelihood phylogenetic trees of the S. ruber M31T-predicted proteins with an archaeal evolutionary history potentially acquired by horizontal gene transfer: (A) sodium-dependent transporter (XGX02982.1); (B) MFS transporter (XGX01653.1); and (C) ABC transporter ATP-binding protein (XGX02353.1). The organism names are followed by the PID of the sequence from NCBI. The S. ruber M31T sequences group with the archaeal sequences (orange) instead of bacterial (blue) or eukaryotic (green) ones.
Methylation patterns were determined by the same microbial genome analysis pipeline (SMRT Link v11.1.0.166339), yielding two m6A methylated DNA motifs: GATANNNNNCTC and CAGCAG. Homology-based searches of the assembled genome identified six restriction-modification (R-M) system genes that were recorded in the REBASE database (18). BLAST analysis was used to identify RM system genes from the complete genome sequence by using each open reading frame in the genome to query NCBI nr and the REBASE databases (18, 19). Multiple sequence alignments were performed using the PROMALS 3D server (20). Type I methylase M.SruM31TI (ACED82_05905) was assigned to the GATANNNNNCTC motif, and no gene was assigned to the CAGCAG motif yet.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Antón J, Oren A, Benlloch S, Rodríguez-Valera F, Amann R, Rosselló-Mora R. 2002. Salinibacter ruber gen. nov., sp. nov., a novel, extremely halophilic member of the bacteria from saltern crystallizer ponds. Int J Syst Evol Microbiol 52:485–491. doi:10.1099/00207713-52-2-48511931160 · doi ↗ · pubmed ↗
- 2Mongodin EF, Nelson KE, Daugherty S, De Boy RT, Wister J, Khouri H, Weidman J, Walsh DA, Papke RT, Sanchez Perez G, Sharma AK, Nesbø CL, et al.. 2005. The genome of Salinibacter ruber: convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc Natl Acad Sci USA 102:18147–18152. doi:10.1073/pnas.050907310216330755 PMC 1312414 · doi ↗ · pubmed ↗
- 3Das Sarma S. 2006. Extreme halophiles are models for astrobiology. Microbe Magazine 1:120–126. https://halo-ed.org/73_Das Sarma_Microbe_2006.pdf
- 4Oren A. 2013. Salinibacter: an extremely halophilic bacterium with archaeal properties. FEMS Microbiol Lett 342:1–9. doi:10.1111/1574-6968.1209423373661 · doi ↗ · pubmed ↗
- 5ATCC. ATCC medium: 2402 Salinibacter ruber medium. ATCC, Manassas, VA, USA. Available from: https://www.atcc.org/-/media/product-assets/documents/microbial-media-formulations/2/4/0/2/atcc-medium-2402.pdf?rev=cdc 4a 1ab 32f 243209 bfa 9b 3e 0d 8e 0534. Retrieved 26 May 2025.
- 6Sambrook J, Russel DW. 2001. Molecular cloning: a laboratory manual. 3rd edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 7Besemer J, Lomsadze A, Borodovsky M. 2001. Gene Mark S: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. doi:10.1093/nar/29.12.260711410670 PMC 55746 · doi ↗ · pubmed ↗
- 8Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi:10.1093/bioinformatics/btu 15324642063 · doi ↗ · pubmed ↗