CaDAVEr: a metagenome-assembled genome catalog of microbial decomposers across vertebrate environments
Valerie A. Seitz, Bridget B. McGivern, Michael Shaffer, Mikayla A. Borton, Aeriel D. Belk, Parsa Ghadermazi, Cameron Martino, Liat Shenhav, Anru R. Zhang, Pixu Shi, Alexandra Emmons, Heather Deel, Zhenjiang Zech Xu, Victoria Nieciecki, Qiyun Zhu, Kalen Cantrell, Asa Ben-Hur

TL;DR
This paper introduces a catalog of microbial genomes from soil near decomposing vertebrates to better understand decomposition processes.
Contribution
The study presents 277 new metagenome-assembled genomes from cadaver-associated soils.
Findings
The genomes were recovered from environments associated with vertebrate decomposition.
These genomes can improve understanding of microbial roles in organic matter degradation.
Abstract
Microbial degradation of organic matter is a fundamental Earth process, yet a mechanistic understanding of microbial metabolisms and successional ecology involved in decomposition remains poorly understood. Here, we announce the recovery of 277 cadaver-associated soil metagenome-assembled genomes to enhance our understanding of vertebrate decomposition microbial processes.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
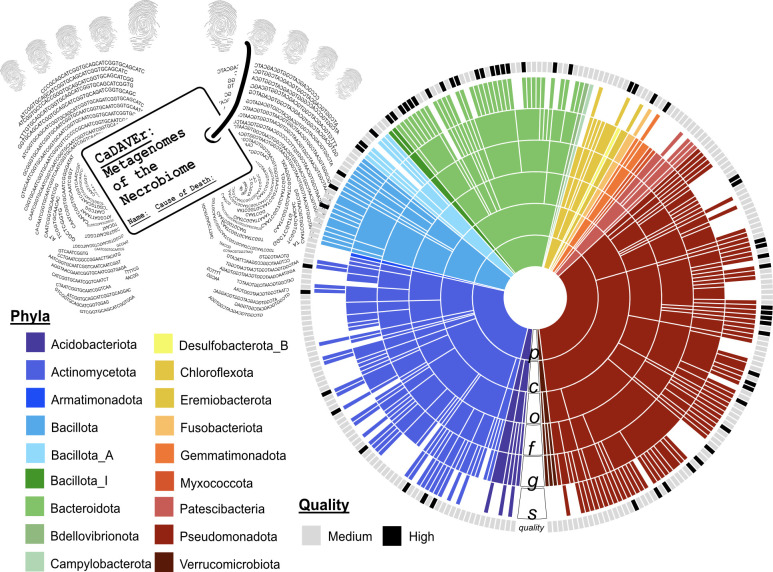 Fig 1
Fig 1| MAG ID | NCBI accession | Genome | Coverage (×) | Number of scaffolds | N50 | Completeness (%) | Contamination (%) | GC | # Predicted genes | Taxonomy (genus) |
|---|---|---|---|---|---|---|---|---|---|---|
| CMU_advanced.final.11 |
| 1,738,034 | 111 | 186 | 12,259 | 89.67 | 0.88 | 44.3 | 1,802 | g__Wohlfahrtiimonas |
| CMU.bins.224 |
| 1,447,706 | 78 | 42 | 84,618 | 81.62 | 0.47 | 29.9 | 1,462 | g__JASLAT01 |
| CMU.bins.248 |
| 1,960,261 | 382 | 271 | 8,493 | 80.27 | 2.29 | 40.5 | 1,759 | g__Ignatzschineria |
| CMU.bins.298 |
| 1,379,843 | 3.71502592 | 260 | 5,412 | 71.93 | 2.18 | 41.6 | 1,309 | g__Ignatzschineria |
| CMU.bins.69 |
| 2,179,327 | 13.2268609 | 266 | 10,819 | 79.66 | 6.9 | 41.2 | 1,933 | g__Ignatzschineria |
| SHSU_active.final.55 |
| 2,230,926 | 31 | 222 | 16,150 | 95.52 | 0.94 | 37.6 | 2,311 | g__Savagea |
| SHSU_active.final.81 |
| 2,756,840 | 55 | 165 | 23,680 | 97.42 | 0 | 33.7 | 2,234 | g__Bacteroides_E |
| SHSU_advanced.final.156 |
| 1,997,474 | 7 | 274 | 9,700 | 90.58 | 2.08 | 37.7 | 1,899 | g__Bacteroides_E |
| SHSU_advanced.final.35 |
| 1,258,874 | 23 | 248 | 5,499 | 74.04 | 1.14 | 38.4 | 1,390 | g__Vagococcus_A |
| SHSU.bins.123 |
| 3,542,934 | 63 | 533 | 7,822 | 82.18 | 4.29 | 51 | 3,625 | g__Morganella |
| SHSU.bins.132 |
| 2,116,720 | 54 | 106 | 25,442 | 99.71 | 1.17 | 45.6 | 1,961 | g__CALZBJ01 |
| SHSU.bins.167 |
| 2,263,551 | 21 | 281 | 10,811 | 88.41 | 1.95 | 33.6 | 2,071 | g__Bacteroides_E |
| SHSU.bins.175 |
| 1,981,783 | 25 | 263 | 9,902 | 78.09 | 6.73 | 41.9 | 1,749 | g__Ignatzschineria |
| SHSU.bins.195 |
| 2,596,507 | 37 | 254 | 15,705 | 88.91 | 1.17 | 39.7 | 2,325 | g__Ignatzschineria |
| SHSU.bins.354 |
| 1,538,193 | 46 | 131 | 14,935 | 94.25 | 0.58 | 45.4 | 1,533 | g__JAAZCI01 |
| SHSU.bins.365 |
| 1,752,486 | 84 | 45 | 63,825 | 97.27 | 0 | 44.1 | 1,629 | g__Ignatzschineria |
| SHSU.bins.50 |
| 4,056,206 | 45 | 729 | 6,174 | 51.88 | 0 | 58 | 4,297 | g__Klebsiella |
| SHSU.bins.517 |
| 1,912,837 | 24 | 134 | 22,260 | 87.43 | 3.8 | 37 | 1,898 | g__Wohlfahrtiimonas |
| SHSU.bins.531 |
| 1,451,374 | 171 | 291 | 5,212 | 62.9 | 3.31 | 40.5 | 1,361 | g__Ignatzschineria |
| SHSU.bins.89 |
| 1,507,431 | 46 | 222 | 8,450 | 60.92 | 4.78 | 38.7 | 1,408 | g__Ignatzschineria |
| UTK_active.final.18 |
| 1,605,292 | 242 | 244 | 7,889 | 77.55 | 0.7 | 47.6 | 1,679 | g__Thiopseudomonas |
| UTK_active.final.26 |
| 1,609,873 | 46 | 254 | 7,156 | 85.83 | 4.17 | 46.1 | 1,799 | g__Savagea |
| UTK.bins.107 |
| 2,578,716 | 47 | 455 | 6,343 | 79.81 | 1.15 | 40.1 | 2,608 | g__Acinetobacter |
| UTK.bins.126 |
| 1,906,363 | 15 | 352 | 5,909 | 71.72 | 9.56 | 40.4 | 1,925 | g__Ignatzschineria |
| UTK.bins.19 |
| 2,219,372 | 43 | 159 | 23,955 | 95.47 | 1.3 | 39.5 | 1,957 | g__Bacteroides_E |
| UTK.bins.42 |
| 1,863,275 | 18 | 142 | 19,613 | 93.6 | 0.39 | 40 | 1,884 | g__Savagea |
| UTK.bins.50 |
| 1,565,609 | 48 | 53 | 63,371 | 96.34 | 0.91 | 36.5 | 1,510 | g__CALAZW01 |
| UTK.bins.71 |
| 1,322,987 | 34 | 287 | 4,565 | 64.9 | 5.35 | 46 | 1,491 | g__Savagea |
| UTK.bins.74 |
| 1,649,164 | 11.0139592 | 25 | 146,272 | 97.95 | 0 | 44.3 | 1,534 | g__JAAZCI01 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEnvironmental DNA in Biodiversity Studies · Forensic Entomology and Diptera Studies · Molecular Biology Techniques and Applications
ANNOUNCEMENT
Decomposition of organic material, including plants, animals, and their byproducts, is one of the most important ecosystem processes on Earth, requiring complex metabolisms and multi-trophic food webs to efficiently recycle nutrients. Animal tissue decomposition is particularly understudied despite its potential role for innovating agricultural and forensic tools. Microbial communities critical to animal tissue decomposition, also known as the necrobiome (1), lack metagenomic data characterizing this metabolism. Using soils longitudinally collected from 36 terrestrially decomposing human cadavers (n = 21 timepoints per donor) across three U.S. locations (Knoxville, TN; Grand Junction, CO; Huntsville, TX), we present a genomic resource for microbial decomposers of animal tissue (2). Here, we introduce 277 medium- and high-quality metagenome-assembled genomes (MAGs) comprising the CaDAVEr (Catalog of microbial Decomposers Across Vertebrate Environments) database. We highlight 29 taxa previously identified as central members to a co-occurrence network associated with advanced decomposition (Table 1) (2). We have previously shown that these microbes are ubiquitous vertebrate decomposers with insects as a likely vector (2). CaDAVEr members are largely undetected in large databases of host-associated or soil microbial communities, supporting the notion that the necrobiome has an untapped suite of microbial community and functional diversity (2).
Swab samples were collected across decomposition stages from soils adjacent to 36 human cadavers (n = 756). Soil controls (n = 9), blank controls (n = 102), and no-template PCR controls (n = 15) were included. DNA was extracted using the PowerSoil DNA isolation kit 96-htp (MoBio Laboratories) and prepared for shallow metagenomic sequencing. Sequencing (2 × 151 bp) was completed on either an Illumina HiSeq 4000 or an Illumina NovaSeq 6000, resulting in 1,271 Gbp of sequencing and 4,211,654,692 total reads. Adapters were removed, and reads were filtered using Atropos (q = 15, --minimum-length 100, v.1.1.24) (3). Bowtie2 (v.2.2.3) (4) was used to align human sequences against the Genome Reference Consortium Human Build 38 patch release 13, removing all data that matched the reference. Resulting filtered SAM files were converted to FASTQ format with samtools (5) (v.1.3.1) and bedtools (6) (v.2.26.0). Samples with <500 k reads were removed from the analysis as a quality control measure, resulting in a final metagenomics data set of 569 hip-adjacent soil samples, five soil controls, and four no-template controls (N = 575).
Using protocols described previously to maximize genome recovery (7, 8), metagenomes were assembled by geographic site and decomposition stage (MEGAHIT, v.1.2.9, –k-min 41) (9) with assembled scaffolds >2,500 kb binned into MAGs (MetaBAT2, v.2.12.1) (10). Bin quality was assessed by checkM (v.1.1.2) (11), retaining only medium- and high-quality MAGs, which resulted in 1,130 total MAGs (>50% estimated completion and <10% contamination). MAGs were then dereplicated at 99% identity (dRep, v.2.6.2) (12), resulting in 277 MAGs in CaDAVEr, of which 24% are considered high quality (Fig. 1). MAGs were assigned taxonomy via GTDB-tk (v.2.4.0, r220) (13), and genes were annotated using DRAM (v.1.0.0) (14) and were predominantly assigned to the phyla Pseudomonadota (dark red, n = 99), Actinomycetota (dark blue, n = 70), and Bacteroidota (light green, n = 41; Fig. 1). The package default parameters were used unless otherwise specified. Resources of the CaDAVEr database broaden the taxonomic and functional characterization of the vertebrate necrobiome, contributing important phylogenetic and genomic information within decomposition.
CaDAVEr yields a robust MAG database of microbial decomposers. The taxonomy of the 277 dereplicated MAGs is shown by sequentially colored rings ordered from phylum (P), class (C), order (O), family (F), genus (G), to species (S) assignment. Ring color corresponds to phyla, with the taxonomic assignment denoted in the legend to the left. Gaps at each level represent MAGs that were unclassified at that level of taxonomy (according to GTDB-tk v.2.4.0, r220). The outer ring shows the MAG quality classification, where gray is a medium-quality MAG (completion >50% and contamination <10%), and black is a high-quality MAG (completion >90% and contamination <10%).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Benbow ME, Lewis AJ, Tomberlin JK, Pechal JL. 2013. Seasonal necrophagous insect community assembly during vertebrate carrion decomposition. J Med Entomol 50:440–450. doi:10.1603/me 1219423540134 · doi ↗ · pubmed ↗
- 2Burcham ZM, Belk AD, Mc Givern BB, Bouslimani A, Ghadermazi P, Martino C, Shenhav L, Zhang AR, Shi P, Emmons A, et al.. 2024. A conserved interdomain microbial network underpins cadaver decomposition despite environmental variables. Nat Microbiol 9:595–613. doi:10.1038/s 41564-023-01580-y 38347104 PMC 10914610 · doi ↗ · pubmed ↗
- 3Didion JP, Martin M, Collins FS. 2017. Atropos: specific, sensitive, and speedy trimming of sequencing reads. Peer J 5:e 3720. doi:10.7717/peerj.372028875074 PMC 5581536 · doi ↗ · pubmed ↗
- 4Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi:10.1038/nmeth.192322388286 PMC 3322381 · doi ↗ · pubmed ↗
- 5Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence Alignment/Map format and SA Mtools. Bioinformatics 25:2078–2079. doi:10.1093/bioinformatics/btp 35219505943 PMC 2723002 · doi ↗ · pubmed ↗
- 6Quinlan AR, Hall IM. 2010. BED Tools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi:10.1093/bioinformatics/btq 03320110278 PMC 2832824 · doi ↗ · pubmed ↗
- 7Mc Givern BB, Cronin DR, Ellenbogen JB, Borton MA, Knutson EL, Freire-Zapata V, Bouranis JA, Bernhardt L, Hernandez AI, Flynn RM, Woyda R, Cory AB, Wilson RM, Chanton JP, Woodcroft BJ, Ernakovich JG, Tfaily MM, Sullivan MB, Tyson GW, Rich VI, Hagerman AE, Wrighton KC. 2024. Microbial polyphenol metabolism is part of the thawing permafrost carbon cycle. Nat Microbiol 9:1454–1466. doi:10.1038/s 41564-024-01691-038806673 PMC 11153144 · doi ↗ · pubmed ↗
- 8Mc Givern BB, Tfaily MM, Borton MA, Kosina SM, Daly RA, Nicora CD, Purvine SO, Wong AR, Lipton MS, Hoyt DW, Northen TR, Hagerman AE, Wrighton KC. 2021. Decrypting bacterial polyphenol metabolism in an anoxic wetland soil. Nat Commun 12:2466. doi:10.1038/s 41467-021-22765-133927199 PMC 8084988 · doi ↗ · pubmed ↗