Draft genome sequences of human adenovirus F associated with acute gastroenteritis in Blantyre, Malawi
Flywell Kawonga, Ernest Matambo, End Chinyama, Chimwemwe Mhango, Clara Majengo, Josephine Msowoya, Benjamin Kumwenda, Celeste M. Donato, Arox W. Kamng'ona, Milton T. Mogotsi, Nkosazana Shange, Ayodeji E. Ogunbayo, Francis E. Dennis, Martin M. Nyaga, Chrispin Chaguza

TL;DR
This paper presents four draft genome sequences of a human adenovirus linked to childhood gastroenteritis in Malawi.
Contribution
The study provides new draft genomes of HAdV-F genotype 40/41 from Malawi, contributing to global understanding of this pathogen.
Findings
Four draft genomes of HAdV-F were sequenced from children with acute gastroenteritis in Blantyre, Malawi.
The viruses belong to genotype 40/41, known to cause pediatric gastroenteritis worldwide.
Abstract
Human adenovirus F (HAdV-F), genotype 40/41, ranks as the second leading cause of pediatric viral gastroenteritis globally. Here, we report four draft genomes of HAdV-F from Malawi, obtained from children with acute gastroenteritis at Queen Elizabeth Central Hospital and Bangwe Health Centre, Blantyre, between 2012 and 2024.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
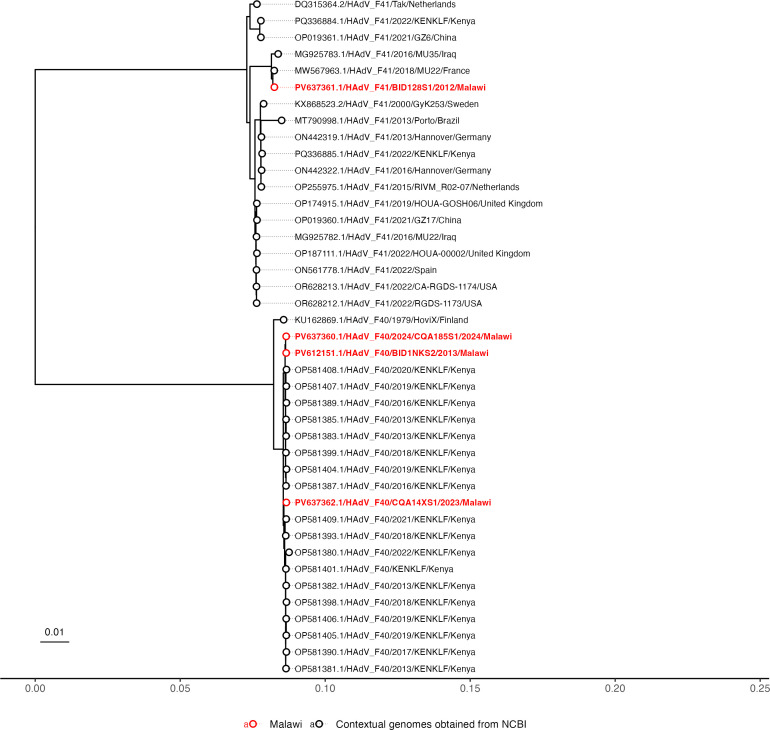 Fig 1
Fig 1| BID128S1 | BID1NKS2 | CQA14XS1 | CQA185S1 | |
|---|---|---|---|---|
| Collection year | 2012 | 2013 | 2023 | 2024 |
| Sample type | Stool | Stool | Stool | Stool |
| Cycle threshold (Ct) | 23.8 | 9.8 | 16.9 | 27 |
| Sequencing platform | NextSeq 2000 | NextSeq 2000 | NextSeq 2000 | NextSeq 2000 |
| Total number of reads generated | 507,336 | 515,370 | 536,530 | 654,632 |
| Total mapped reads to the reference | 296,038 | 267,408 | 93,055 | 529,873 |
| Raw sequence length | 36–151 | 36–151 | 36–151 | 36–151 |
| Genome length | 34,013 | 34,207 | 34,204 | 34,204 |
| GC (%) | 50 | 51 | 52 | 49 |
| Genome depth | 972.186× | 1027.25× | 335.299× | 1951.61× |
| Reference nucleotide identity (%) | 99.8 | 99.7 | 99.7% | 99.7% |
| CDS | 35 | 30 | 31 | 31 |
| Genotype | HAdV-F41 | HAdV-F40 | HAdV-F40 | HAdV-F40 |
| GenBank accession number |
|
|
|
|
- —Gates Foundationhttp://dx.doi.org/10.13039/100000865
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVirus-based gene therapy research · Viral gastroenteritis research and epidemiology · Bacteriophages and microbial interactions
ANNOUNCEMENT
Human adenovirus F (HAdV-F), genotypes 40 and 41 are major causes of pediatric viral gastroenteritis, accounting for 2.8–11.8% of diarrheal cases in infants worldwide (1). The HAdV belongs to the Mastadenovirus faecale genus within the family Adenoviridae. The average genome sizes of HAdV-F40 and HAdV-F41 are 34,214 and 34,188 base pairs, respectively (2, 3). In Malawi, HAdV-F (genotype 40/41) was the second most frequently detected pathogen associated with diarrhea, identified in 29.1% of cases at Queen Elizabeth Central Hospital (QECH) in our previous study (4). No genomic studies of HAdV-F have been conducted in Malawi. This report presents four draft genome sequences of HAdV-F from children under 5 years old with acute gastroenteritis at QECH and Bangwe Health Centre, as part of the Sequencing and Antigenic Cartography of Enteric Viruses (SACEV) project.
Ten samples per month were screened by polymerase chain reaction (PCR) using customized TaqMan Array Cards as previously described (5). Four positive adenovirus samples with PCR cycle threshold (Ct) <35 were selected for whole genome sequencing. Nucleic acid was extracted from adenovirus-positive stool samples using QIAamp Fast DNA Stool mini kit (Qiagen, Hilden, Germany). DNA extracts were quantified using a High Sensitivity dsDNA Assay on a Qubit Flex Fluorometer (Thermo Fisher Scientific, USA).
The HAdV-F described here was generated by shotgun metagenomic sequencing. Extracted DNA was subjected to whole transcriptome amplification using a Qiagen FX Whole Transcriptome Amplification kit, generating genomic sequences of all the genetic material present in the sample. Genomic libraries were prepared using the Illumina DNA Prep kit (Illumina, USA) before being sequenced on the Illumina NextSeq 2000 platform using a P1 flow cell and 300-cycle reagent kit (2 × 150 bp paired-end reads). Quality control was performed using FastQC 0.11.7 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Low-quality reads were trimmed using Trimmomatic (v0.39) (6). To remove human reads, the trimmed sequences were aligned to the human genome (Homo_sapiens.GRCh38.dna.primary_assembly.fa, NCBI accession ID: GCA_000001405.15) using Bowtie2 2.5.4 with default parameters (7). The non-human reads were mapped to NC_001454.1 reference genome using the Burrows-Wheeler Alignment 0.7.18-r1243-dirty with default parameters (8). Consensus genomes were generated by calling high-confidence bases from aligned sequence reads using iVar (v1.4.4) with default parameters (9). The whole-genome assemblies were further annotated using Prokka 1.14.6 (10). The genome assemblies are considered drafts due to unconfirmed 5′ and 3′ terminal sequences, although all major coding regions were successfully recovered. BLASTn of the assembled genomes showed that BID128S1 had 99.89% nucleotide similarity to MK962807.1, whereas BID1NKS2, CQA14XS1, and CQA185S1 had 99.77%, 99.71% and 99.76% similarity to NC_001454.1, respectively. The depth of the genome assemblies was checked using samtools 1.21 (https://www.htslib.org/). The reads and assembly characteristics are summarized in Table 1.
The assembled genomes were identified as human mastadenovirus F using the Genome Detective Virus Tool (https://doi.org/10.1093/bioinformatics/bty695). Genotyping identified BID1NKS2, CQA14XS1, and CQA185S1 as HAdV-F40, and BID128S1 as HAdV-F41, based on the highest sequence similarity from BLASTn analysis. Figure 1 shows the phylogeny of Malawian sequences alongside global contextual genomes, with the Malawian genomes clustering closely with sequences from Kenya and France.
Contextual genomes of HAdV-40 and HAdV-41 were retrieved from the NCBI Virus database, selected based on complete coding sequences and broad global representation. Multiple sequence alignment was performed using MAFFT v7 with default parameters, and a maximum likelihood phylogenetic tree was inferred using IQ-TREE V2.4.0 with the GTR + G substitution model. Branch support was assessed using 1,000 ultrafast bootstrap replicates. The resulting tree was imported into R using the read.tree() function from the ape package (11) and rooted at the Psittacine aviadenovirus C (NCBI Accession: NC_075452.1) using the root() function to establish evolutionary directionality. For clarity, the outgroup was removed using the drop.tip() function from the same package. Strain metadata are used to annotate phylogenetic tree tips, distinguishing Malawian sequences from contextual genomes from NCBI. Malawian strains are shown in red, and contextual genomes are shown as black circles. The tips display the accession numbers, strain names, year of detection, and country of detection.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chandra P, Lo M, Mitra S, Banerjee A, Saha P, Okamoto K, Deb AK, Ghosh SK, Manna A, Dutta S, Chawla-Sarkar M. 2021. Genetic characterization and phylogenetic variations of human adenovirus-F strains circulating in eastern India during 2017-2020. J Med Virol 93:6180–6190. doi:10.1002/jmv.2713634138479 · doi ↗ · pubmed ↗
- 2Lemiale F, Haddada H, Nabel GJ, Brough DE, King CR, Gall JGD. 2007. Novel adenovirus vaccine vectors based on the enteric-tropic serotype 41. Vaccine (Auckl) 25:2074–2084. doi:10.1016/j.vaccine.2006.11.025PMC 258466717250935 · doi ↗ · pubmed ↗
- 3van Loon AE, Ligtenberg M, Reemst AM, Sussenbach JS, Rozijn TH. 1987. Structure and organization of the left-terminal DNA regions of fastidious adenovirus types 40 and 41. Gene 58:109–126. doi:10.1016/0378-1119(87)90034-52961652 · doi ↗ · pubmed ↗
- 4Iturriza-Gómara M, Jere KC, Hungerford D, Bar-Zeev N, Shioda K, Kanjerwa O, Houpt ER, Operario DJ, Wachepa R, Pollock L, Bennett A, Pitzer VE, Cunliffe NA. 2019. Etiology of diarrhea among hospitalized children in Blantyre, Malawi, following rotavirus vaccine introduction: a case-control study. J Infect Dis 220:213–218. doi:10.1093/infdis/jiz 08430816414 PMC 6581894 · doi ↗ · pubmed ↗
- 5Liu J, Gratz J, Amour C, Nshama R, Walongo T, Maro A, Mduma E, Platts-Mills J, Boisen N, Nataro J, Haverstick DM, Kabir F, Lertsethtakarn P, Silapong S, Jeamwattanalert P, Bodhidatta L, Mason C, Begum S, Haque R, Praharaj I, Kang G, Houpt ER. 2016. Optimization of quantitative PCR methods for enteropathogen detection. P Lo S One 11:e 0158199. doi:10.1371/journal.pone.015819927336160 PMC 4918952 · doi ↗ · pubmed ↗
- 6Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi:10.1093/bioinformatics/btu 17024695404 PMC 4103590 · doi ↗ · pubmed ↗
- 7Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi:10.1038/nmeth.192322388286 PMC 3322381 · doi ↗ · pubmed ↗
- 8Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi:10.1093/bioinformatics/btp 32419451168 PMC 2705234 · doi ↗ · pubmed ↗