Clinical characteristics, genetic spectrum and therapeutic effects of 51 male patients with idiopathic hypogonadotropic hypogonadism from southern China
Huiying Sheng, Cuili Liang, Jing Cheng, Huazhen Liu, Xiaojian Mao, Xiuzhen Li, Duan Li, Zhikun Lu, Yanna Cai, Xueying Su, Liyu Zhang, Wen Fu, Jinhua Hu, Wei Jia, Guochang Liu, Wen Zhang, Li Liu, Yunting Lin

TL;DR
This study examines the clinical and genetic features of 51 male IHH patients in southern China and evaluates treatment outcomes.
Contribution
The study expands the genetic spectrum of IHH and identifies clinical markers for early diagnosis and differential diagnosis.
Findings
FGFR1 is the most common causative gene in southern Chinese male IHH patients.
ANOS1 and CHD7 variants show distinct clinical and biochemical profiles.
Treatment significantly improves penile length and hormone levels in IHH patients.
Abstract
Idiopathic hypogonadotropic hypogonadism (IHH) is a set of rare diseases characterized by abnormal sexual development with clinical heterogeneity and genotypic complexity. This study aims to investigate the phenotypic and genotypic characteristics of male IHH in southern China, and evaluate the therapeutic effects of current treatments. Fifty-one male IHH patients from southern China were enrolled in this study. Their clinical, imaging, hormonal and genetic findings were analyzed retrospectively. In this study, the most common causative gene of IHH was FGFR1 (45.10%), followed by ANOS1 (21.57%) and CHD7 (17.65%). Forty-five different variants, including 22 known and 23 novel variants, were found. The mean age at diagnosis was 7.84 ± 5.89 years, the most common clinical phenotype was micropenis (98.04%), the most frequent imaging feature was abnormal ultrasound of sexual glands…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
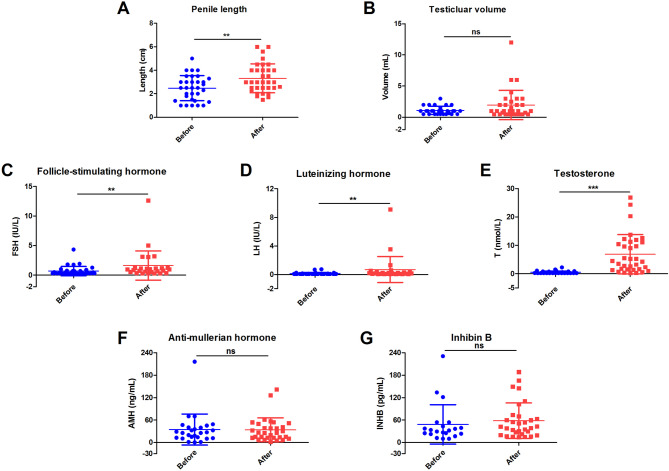 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital Ear and Nasal Anomalies · Hypothalamic control of reproductive hormones · Olfactory and Sensory Function Studies
Introduction
Idiopathic hypogonadotropic hypogonadism (IHH), also known as isolated hypogonadotropic hypogonadism or isolated gonadotropin-releasing hormone (GnRH) deficiency, is a set of rare diseases caused by defects in the hypothalamic-pituitary-gonadal axis, resulting in insufficient GnRH but normal levels of other pituitary hormones [1, 2]. The prevalence of IHH is estimated to be 1–10 per 100,000 births with males having a 2.6-fold to 5-fold higher prevalence than females [3–6].
The typical clinical manifestations of IHH include lack of sexual development, small genitalia, small sexual glands and delayed puberty, accompanying by biochemical features of inappropriately low levels of gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), and sex steroids [3, 7]. According to the olfactory function, IHH can be divided into two groups: (1) Kallmann syndrome, associated with anosmia or hyposmia and accounting for 60% of patients; and (2) normosmic IHH, responsible for the remaining 40% of patients [1, 3].
Clinical variability of expression and penetrance has been observed in patients with IHH due to the genetic heterogeneity [1, 8]. The molecular mechanisms underlying IHH are diverse. To date, more than 30 different causative genes have been reported to cause IHH with inheritance patterns including X-linked recessive, autosomal dominant and autosomal recessive [9–11]. Among them, ANOS1, CHD7, FGFR1, GNRHR, PROK2, PROKR2, IL17RD, SOX10, TAC3, TACR3, FGF8, KISS1 and KISS1R gene are the most frequent molecular bases [3, 9]. However, a genetic etiology has been identified in only about 50% of IHH cases, whereas the cause remains unknown in the others [2, 4, 8, 12].
The treatment of IHH includes steroid replacement therapy, human chorionic gonadotropin (HCG) therapy, combined gonadotropin therapy, pulsatile GnRH therapy, or sequential gonadotropin therapy [9, 13, 14]. Traditionally, patients with IHH are typically not diagnosed until late adolescence or early adulthood due to absence of pubertal development [12]. With advances in genetic testing, especially next-generation sequencing (NGS), the precise molecular diagnosis of IHH patients is facilitated, enabling timely treatment and improving drug response [7, 13].
In the present study, we retrospectively analyzed the clinical, imaging, hormonal and genetic findings of 51 male patients with IHH from southern China to study their clinical characteristics and genetic spectrum, and assess the therapeutic effects of current treatments.
Materials and methods
Patients
Fifty-one male IHH patients from 48 unrelated families were enrolled in Guangzhou Women and Children’s Medical Center from November 2015 to December 2024. All subjects were of Han ethnicity and from southern China. None of the parents were consanguineous.
The inclusion criteria were as follows:1) clinical signs of hypogonadism (including micropenis and/or cryptorchidism) for boys younger than 14 years of age; clinical signs of hypogonadism and no development of secondary sexual characteristics for boys older than 14 years of age; 2) low levels of gonadotropins (LH < 1.0 IU/L) and/or testosterone (< 3.5 nmol/L) without abnormalities of other pituitary and adrenal hormones; 3) normal chromosome karyotype; 4) identification of pathogenic or likely pathogenic variants in the causative genes of IHH [15]. Only patients who meet all four criteria were included in the IHH cohort.
Clinical information
Medical history and clinical data were collected and evaluated by clinicians. Physical examinations were performed by physicians. Family history was obtained by genetic counselors. Serum hormones and chromosome karyotypes were detected in the hospital’s genetics and endocrinology laboratory. Skeletal X-ray and pituitary magnetic resonance imaging (MRI) were conducted in the hospital’s medical imaging center. Ultrasonography of sexual glands was performed in the hospital’s ultrasonic department.
Molecular analysis
All patients and their parents were subjected to molecular analysis to identify the genetic basis of IHH. Genomic DNA (gDNA) was extracted from peripheral blood samples using DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany). For patients enrolled before January 2018, their gDNA samples were first subjected to ANOS1 gene analysis using Sanger sequencing. Subsequently, whole exome sequencing (WES) was performed for ANOS1-negative samples. For patients enrolled after January 2018, their gDNA samples were directly subjected to WES.
The gnomAD was employed to exclude the polymorphic alleles, and HGMD (Professional 2025.2) was engaged to confirm the known pathogenic variant. For novel variants, in-silico tools were used to predict the functional consequences, including PROVEAN, SIFT, PolyPhen-2, MutationTaster, FATHMM, NetGene2, NNSPLICE 0.9 and RNA Splicer. The pathogenicity of variants was evaluated according to the guidelines of American College of Medical Genetics (ACMG) [16].
Treatment and follow-up
After diagnosis, all patients, except those lost to follow-up or newly diagnosed, were treated with intramuscular injection of HCG (500–1000 U) alone or in combination with human menopausal gonadotropin (HMG, 75 U) twice weekly for 6–12 weeks annually. To promote penile growth and orchiocatabasis, or to maintain secondary sex characteristics, some patients were treated with oral testosterone undecanoate (TU) at a dose of 2–3 mg/(kg·d). Only 3 patients older than 14 years of age were transitioned to pulsatile injection of GnRH (5–10 µg/90 min) via an infusion pump.
Most patients were followed up at intervals of 3–12 months, except for a few with poor compliance. The height, weight, penile length, testicular volume and related hormonal parameters were measured at every visit. The doses of medication were adjusted based on the hormonal results.
Statistical analysis
SPSS Statistics 17.0 software (SPSS, Chicago, United States) was used to calculate means and standard deviation (SD), compare means, and evaluate differences. Student’s t test or One-Way ANOVA was performed to compare means for data distributed normally, whereas non-parametric Mann-Whitney U test or Kruskal-Wallis H test was engaged for data not. For cross-tables, Yates’ continuity correction or Fisher’s exact test was conducted where applicable. A statistically significant difference was defined as p < 0.05. The vertical scatter plots showing mean with SD were generated by GraphPad Prism 5 software (GraphPad, Boston, USA).
Results
Mutational spectrum
A total of 51 patients from 48 unrelated families in southern China, including three pairs of siblings, were confirmed to have pathogenic or likely pathogenic variants in the causative genes of IHH. As shown in Table 1 and Table S1, variants in ANOS1, CHD7, FGFR1, HS6ST1, KISS1R, PROKR2 and SOX11 gene accounted for 11 (11/51, 21.57%), 9 (9/51, 17.65%), 23 (23/51, 45.10%), 1 (1/51, 1.96%), 1 (1/51, 1.96%), 3 (3/51, 5.88%) and 3 (3/51, 5.88%) cases, respectively. Among them, FGFR1 gene was the most frequent cause, followed by ANOS1 and CHD7 gene.
Table 1. Deleterious variants identified in 51 male IHH patientsFamilyPatientGenderGeneVariantNucleotide changeAmino acid changeZygosityInheritanceReported previously?ACMG categoryF1P1Male FGFR1 c.797delCinsTT(p.Thr266Ilefs6)Small indelFrameshiftHetPaternalNovel 1PathogenicF2P2Male FGFR1 c.376delG(p.Glu126Argfs26)Small deletionFrameshiftHet De novo Novel 2PathogenicF3P3Male CHD7 c.5222G > C(p.Arg1741Pro)Base substitutionMissenseHet De novo Known 1PathogenicF4P4Male FGFR1 8p11.23-p11.22(36788433–38458282)delGross deletionNAHet De novo Novel 3PathogenicF5P5Male FGFR1 c.424_425delGA(p.Asp142*)Small deletionNonsenseHet De novo Novel 4PathogenicF6P6Male CHD7 c.5050G > A(p.?)Base substitutionSplicingHet De novo Known 2PathogenicF7P7Male PROKR2 c.533G > C(p.Trp178Ser)Base substitutionMissenseHetPaternalKnown 3Likely pathogenicF8P8Male CHD7 c.4186–2 A > GBase substitutionSplicingHet De novo Novel 5PathogenicF9P9Male FGFR1 8p11.23-p11.22(38147917–38975686)delGross deletionNAHet De novo Novel 6PathogenicF10P10Male CHD7 c.3655 C > T(p.R1219*)Base substitutionNonsenseHet De novo Known 4PathogenicF11P11Male CHD7 c.4291 A > C(p.Lys1431Gln)Base substitutionMissenseHet De novo Novel 7Likely pathogenicF12P12Male FGFR1 c.709G > A(p.Gly237Ser)Base substitutionMissenseHet De novo Known 5PathogenicF13P13Male SOX11 c.347 A > G(p.Tyr116Cys)Base substitutionMissenseHet De novo Known 6PathogenicF14P14Male ANOS1 c.814 C > T(p.Arg272*)Base substitutionNonsenseHemiMaternalKnown 7PathogenicF15P15Male FGFR1 c.481 A > G(p.Met161Val)Base substitutionMissenseHet De novo Novel 8Likely pathogenicF16P16Male CHD7 c.3754T > C(p.Cys1252Arg)Base substitutionMissenseHet De novo Novel 9Likely pathogenicF17P17Male FGFR1 c.2025delG(p.Ile676Serfs38)Small deletionFrameshiftHet De novo Novel 10PathogenicF18P18Male FGFR1 c.246_247delAG(p.Glu84Glyfs26)Small deletionFrameshiftHet De novo Known 8PathogenicF19P19Male FGFR1 c.568T > G(p.Trp190Gly)Base substitutionMissenseHet De novo Novel 11Likely pathogenicF20P20Male ANOS1 c.1267 C > T(p.Arg423*)Base substitutionNonsenseHemiMaternalKnown 9PathogenicF21P21Male CHD7 c.5405-7G > ABase substitutionSplicingHet De novo Known 10PathogenicF22P22Male ANOS1 c.209delA(p.?)Small deletionSplicingHemiMaternalNovel 12PathogenicF23P23Male SOX11 c.87 C > A(p.Cys29*)Base substitutionNonsenseHet De novo Known 11PathogenicF24P24Male ANOS1 c.531_541 + 2delTCTGTACAAAGGTSmall deletionSplicingHemiMaternalNovel 13PathogenicF25P25Male FGFR1 c.570G > T(p.Trp190Cys)Base substitutionMissenseHetMaternalNovel 14Likely pathogenicF26P26Male FGFR1 c.2197 A > T(p.Met733Leu)Base substitutionMissenseHet De novo Novel 15Likely pathogenicF27P27Male HS6ST1 c.1144 C > T(p.Arg382Trp)Base substitutionMissenseHetMaternalKnown 12Likely pathogenicF28P28Male FGFR1 c.936G > A(p.Lys312=)Base substitutionSplicingHet De novo Known 13PathogenicF29P29Male KISS1R c.182 C > A(p.Ser61*)Base substitutionNonsenseHetMaternalKnown 14PathogenicF30P30Male FGFR1 c.565 C > T(p.Arg189Cys)Base substitutionMissenseHetPaternalKnown 15Likely pathogenicF31P31Male ANOS1 c.1267 C > T(p.Arg423*)Base substitutionNonsenseHemiMaternalKnown 9PathogenicF32P32Male PROKR2 c.991G > A(p.Val331Met)Base substitutionMissenseHomParentalKnown 16Likely pathogenicF33P33Male ANOS1 c.1503_1506delTGTC(p.Val502Asnfs46)Small deletionFrameshiftHemiMaternalNovel 16PathogenicF34P34Male FGFR1 c.1411_1414delTGGGinsCCC(p.Trp471Profs10)Small indelFrameshiftHet De novo Novel 17PathogenicF35P35Male FGFR1 c.11G > A(p.Trp4*)Base substitutionNonsenseHetMaternalKnown 17PathogenicF36P36Male SOX11 c.158T > G(p.Met53Arg)Base substitutionMissenseHet De novo Known 18Likely pathogenicF37P37Male FGFR1 c.1828G > C(p.Gly610Arg)Base substitutionMissenseHet De novo Novel 18Likely pathogenicF38P38Male CHD7 c.7831–2 A > GBase substitutionSplicingHetPaternalNovel 19PathogenicF39P39Male FGFR1 c.709G > A(p.Gly237Ser)Base substitutionMissenseHet De novo Known 5PathogenicF33P40Male ANOS1 c.1503_1506delTGTC(p.Val502Asnfs46)Small deletionFrameshiftHemiMaternalNovel 16PathogenicF40P41Male FGFR1 c.1429delA(p.?)Small deletionSplicingHet De novo Novel 20PathogenicF41P42Male ANOS1 c.668G > A(p.Trp223)Base substitutionNonsenseHemiMaternalNovel 21PathogenicF42P43Male CHD7 c.3226 A > G(p.Lys1076Glu)Base substitutionMissenseHet De novo Novel 22Likely pathogenicF43P44Male FGFR1 c.1780 C > T(p.Gln594*)Base substitutionNonsenseHet De novo Known 19PathogenicF24P45Male ANOS1 c.531_541 + 2delTCTGTACAAAGGTSmall deletionSplicingHemiMaternalNovel 13PathogenicF44P46Male PROKR2 c.533G > C(p.Trp178Ser)Base substitutionMissenseHetPaternalKnown 3Likely pathogenicF45P47Male FGFR1 c.2084 C > T(p.Thr695Ile)Base substitutionMissenseHet De novo Known 20PathogenicF46P48Male FGFR1 c.817G > A(p.Val273Met)Base substitutionMissenseHetMaternalKnown 21Likely pathogenicF47P49Male FGFR1 c.1049 C > T(p.Ser350Phe)Base substitutionMissenseHet De novo Novel 23Likely pathogenicF48P50Male ANOS1 c.784 C > T(p.Arg262*)Base substitutionNonsenseHemiMaternalKnown 22PathogenicF48P51Male ANOS1 c.784 C > T(p.Arg262*)Base substitutionNonsenseHemiMaternalKnown 22PathogenicHet, heterozygous; Hemi, hemizygous; Hom, homozygous
Of the 51 IHH patients, 39 (39/51, 76.47%) carried a heterozygous variant, 11 (11/51, 21.59%) harbored a hemizygous variant, and only 1 (1/51, 1.96%) had a homozygous variant. De novo deleterious variants accounted for more than half of cases (29/51, 56.86%), whereas maternal and paternal variants were responsible for 31.37% (16/51) and 9.80% (5/51) of cases respectively, and only 1 patient (1/51, 1.96%) had a homozygous variant transmitted from both parents.
Forty-five different variants, including 22 known and 23 novel variants, were disclosed without a hotspot variant. Among them, 33 (33/45, 73.33%) were base substitutions, 8 (8/45, 17.78%) were small deletions, 2 (2/45, 4.44%) were gross deletions and 2 (2/45, 4.44%) were small indels according to the nucleotide change. When corresponding to the amino acid change, 19 (19/45, 42.22%) were missense variants, 10 (10/45, 22.22%) were nonsense variants, 8 (8/45, 17.78%) altered the splicing site, 7 (7/45, 15.56%) arose to a frameshift effect, and the rest 2 gross deletions (2/45, 4.44%) were denoted as NA because it is difficult to define their roles in message RNA processing and protein translation.
Clinical characteristics at diagnosis
The clinical, imaging and hormonal findings of 51 IHH patients at diagnosis are displayed in Table S2, Table S3 and Table S4 respectively, and summarized in Table 2.
Table 2. Clinical, imaging and hormonal findings of 51 male IHH patients at diagnosis (grouped by age at diagnosis)CharacteristicTotal patients (n = 51)Patients < 14 years (n = 41)Patients ≥ 14 years (n = 10)p value^a^Family history13.73% (7/51)9.76% (4/41)30.00% (3/10)0.248Age at diagnosis (years)7.84 ± 5.89 (n = 51)5.90 ± 4.81 (n = 41)15.76 ± 1.68 (n = 10)0.000Height (SDS)-0.93 ± 1.45 (n = 37)-0.99 ± 1.37 (n = 28)-0.74 ± 1.76 (n = 9)0.666Weight (SDS)-0.71 ± 1.66 (n = 46)-0.92 ± 1.51 (n = 36)0.07 ± 2.02 (n = 10)0.095Penile length (cm)2.41 ± 1.03 (n = 45)2.17 ± 0.92 (n = 36)3.39 ± 0.86(n = 10)0.001Testicular volume (mL)1.12 ± 0.63 (n = 41)0.97 ± 0.53 (n = 31)1.58 ± 0.73(n = 10)0.013Clinical features Growth retardation (height < -2 SD)24.32% (9/37)21.43% (6/28)33.33% (3/9)0.657 Micropenis98.04% (50/51)100.00% (41/41)90.00% (9/10)0.196 Cryptorchidism49.02% (25/51)51.22% (21/41)40.00% (4/10)0.777 Secondary sexual characteristics0.00% (0/51)0.00% (0/41)0.00% (0/10)NA Impaired olfactory function37.93% (11/29)36.84% (7/19)40.00% (4/10)1.000 Facial anomaly11.76% (6/51)12.20% (5/41)10.00% (1/10)1.000 Ocular abnormality9.80% (5/51)12.20% (5/41)0.00% (0/10)0.569 Hearing loss9.80% (5/51)12.20% (5/41)0.00% (0/10)0.569 Laryngeal problems3.92% (2/51)4.88% (2/41)0.00% (0/10)1.000 Congenital heart disease11.76% (6/51)14.63% (6/41)0.00% (0/10)0.331 Gastroesophageal reflux1.96% (1/51)2.44% (1/41)0.00% (0/10)1.000 Unilateral absence of kidney3.92% (2/51)2.44% (1/41)10.00% (1/10)0.357 Skeletal issues3.92% (2/51)4.88% (2/41)0.00% (0/10)1.000 Developmental delay11.76% (6/51)14.63% (6/41)0.00% (0/10)0.331 Weakness1.96% (1/51)2.44% (1/41)0.00% (0/10)1.000 Enuresis1.96% (1/51)0.00% (0/41)10.00% (1/10)0.196Imaging findings Delayed bone age61.90% (13/21)46.15% (6/13)87.50% (7/8)0.085 Abnormal ultrasound of sexual glands86.84% (33/38)82.79% (24/29)100.00% (9/9)0.312 Abnormal pituitary MRI47.06% (8/17)22.22% (2/9)75.00% (6/8)0.057Gonadal hormones^b^ Basal FSH (IU/L)0.80 ± 0.77 (n = 51)0.84 ± 0.82 (n = 41)0.62 ± 0.52 (n = 10)0.418 Basal LH (IU/L)0.16 ± 0.27 (n = 51)0.17 ± 0.29 (n = 41)0.12 ± 0.09 (n = 10)0.662 Basal T (nmol/L)0.46 ± 0.39 (n = 51)0.37 ± 0.25 (n = 41)0.83 ± 0.61 (n = 10)0.000 Basal AMH (ng/mL)32.42 ± 35.52 (n = 41)33.64 ± 37.51 (n = 34)26.51 ± 24.94 (n = 7)0.635 Basal INHB (pg/mL)39.59 ± 42.81 (n = 36)42.85 ± 46.08 (n = 30)23.30 ± 11.49 (n = 6)0.314 Basal LH < 1.0 IU/L98.04% (50/51)97.56% (40/41)100.00% (10/10)1.000 Basal T < 3.5 nmol/L100.00% (51/51)100.00% (41/41)100.00% (10/10)NA Low basal AMH48.79% (20/41)55.89% (19/34)14.29% (1/7)0.093 Low basal INHB41.67% (15/36)30.00% (9/30)100.00% (6/6)0.003 Peak FSH (IU/L) after GnRH stimulation2.60 ± 2.47 (n = 11)1.56 ± 1.87 (n = 3)2.99 ± 2.66 (n = 8)0.421 Peak LH (IU/L) after GnRH stimulation1.61 ± 2.29 (n = 11)0.49 ± 0.53 (n = 3)2.03 ± 2.58 (n = 8)0.346 LH/FSH after GnRH stimulation0.53 ± 0.34 (n = 11)0.36 ± 0.05 (n = 3)0.59 ± 0.38 (n = 8)0.838 T after HCG stimulation (nmol/L)2.10 ± 2.89 (n = 25)2.31 ± 3.19 (n = 20)1.24 ± 0.80 (n = 5)0.838 AMH after HCG stimulation (ng/mL)30.73 ± 20.74 (n = 23)29.25 ± 20.74 (n = 19)37.75 ± 22.20 (n = 4)0.469 INHB after HCG stimulation (pg/mL)41.80 ± 24.04 (n = 21)41.89 ± 25.17 (n = 17)41.41 ± 21.74 (n = 4)0.972SDS, standard deviation score; FSH, follicle-stimulating hormone; LH, luteinizing hormone; T, testosterone; AMH, anti-mullerian hormone; INHB, inhibin B; GnRH, gonadotropin-releasing hormone^a^The p value comparing means between < 14y and ≥ 14y group was calculated using student’s t test or Mann-Whitney U test. The p value of four-fold table was calculated using Yates’ continuity correction or Fisher’s exact test^b^Those gonadal hormones below the lower detectable limit were valued with the lower detectable limit
Of these 51 patients, 7 (7/51, 13.46%) had a family history. Their age at diagnosis varied from 0.25 to 19.75 years with a mean age of 7.84 ± 5.89 years. The mean height standard deviation score (SDS) and weight SDS were − 0.93 ± 1.45 and − 0.71 ± 1.66, respectively [17]. The majority of patients (28/37, 75.68%) had a normal height, whereas 24.32% (9/37) of cases underwent growth retardation with a height below the − 2 SD of the pediatric reference height range.
In our IHH cohort, the most common clinical feature was micropenis (50/51, 98.04%), followed by cryptorchidism (25/51, 49.02%) and impaired olfactory function (11/29, 37.93%). None of the patients had hypospadias. Despite 19.61% (10/51) of patients being older than 14 years of age, none developed secondary sexual characteristics. The mean penile length and testicular volume of our patients were 2.41 ± 1.03 cm and 1.12 ± 0.63 mL, respectively. Additional clinical signs were presented in a few patients, including facial anomaly, ocular abnormality, hearing loss, laryngeal problems, congenital heart disease, gastroesophageal reflux, unilateral absence of kidney, skeletal issues, developmental delay, weakness and enuresis.
Skeletal X-ray, ultrasonography of sexual glands and pituitary MRI were conducted in 21, 38 and 17 patients, respectively. The most frequent imaging feature was abnormal ultrasound of sexual glands (33/38, 86.84%) with bilateral inguinal testes and small testes being the most common. Delayed bone age was revealed in 61.90% of patients (13/21), while pituitary MRI findings were shown in 47.06% of patients (8/17). Notably, only morphological changes or cysts rather than substantive lesions were found by pituitary MRI as the growth hormone, thyroid-stimulation hormone, prolactin, and adrenocorticotrophic hormone of these patients were normal (data not shown).
In basal conditions, the mean hormone levels were as follows: FSH, 0.80 ± 0.77 IU/L; LH, 0.16 ± 0.27 IU/L; testosterone, 0.46 ± 0.39 nmol/L; anti-mullerian hormone (AMH), 32.42 ± 35.52 ng/mL; inhibin B (INHB), 39.59 ± 42.81 pg/mL. Most cases (50/51, 98.04%) had a low LH (< 1.0 IU/L), and all (51/51, 100.00%) had a low testosterone (< 3.5 nmol/L). According to the age-related reference ranges, low AMH and INHB were shown in 48.79% (20/41) and 41.67% (15/36) of patients respectively.
In this retrospective study, 11 patients were subjected to GnRH stimulation test. The peak levels of FSH and LH were 2.60 ± 2.47 IU/L and 1.61 ± 2.29 IU/L with a LH/FSH ratio of 0.53 ± 0.34. Moreover, HCG stimulation test was performed in 25 patients, resulting in an increase of testosterone to 2.10 ± 2.89 nmol/L, whereas AMH and INHB remained similar to baseline with a value of 30.73 ± 20.74 ng/mL and 41.80 ± 24.04 pg/mL, respectively.
Age-phenotype correlation
Based on the age at diagnosis, the 51 patients were categorized into two groups: (1) “< 14 years” group, accounting for the majority of patients (41/51, 80.39%); (2) “≥ 14 years” group, including the remaining 10 patients (10/51, 19.61%).
Significant differences were identified in penile length, testicular volume, basal testosterone, and the proportion of patients with low basal INHB between the two groups (Table 2).
Genotype-phenotype correlation
According to the molecular spectrum, the 51 patients were divided into 4 groups: (1) “ANOS1” group, consisting of 11 patients carrying an ANOS1 variant; (2) “CHD7” group, involving 9 patients having a CHD7 variant; (3) “FGFR1” group, comprising 23 patients with a FGFR1 variant; (4) “other” group, including 1, 1, 3 and 3 patients harboring a HS6ST1, KISS1R, PROKR2 and SOX11 variant respectively, totaling 8 patients.
Despite distinct molecular bases, all groups shared typical features of micropenis and absence of secondary sexual characteristics, and exhibited comparable penile lengths and testicular volumes. Genotype-phenotype correlations were also observed: (1) Most ANOS1 patients had a family history, impaired olfactory function, and significantly lower basal AMH. Except for unilateral absence of kidney in 2 cases, no other accompanying phenotypes were observed in ANOS1 patients; (2) CHD7 patients were younger at diagnosis, but the difference was not significant. CHARGE features, including growth retardation, facial anomaly, ocular abnormality, hearing loss and congenital heart disease [21], and higher basal FSH and LH appeared more frequently in CHD7 patients (Table 3).
Table 3. Clinical, imaging and hormonal findings of 51 male IHH patients at diagnosis (grouped by genotype)CharacteristicANOS1 patients (n = 11)CHD7 patients (n = 9)FGFR1 patients (n = 23)Other patients (n = 8)p value^a^Family history54.55% (6/11)0.00% (0/9)4.35% (1/23)0.00% (0/8)0.000Age at diagnosis (years)10.87 ± 5.64 (n = 11)4.16 ± 5.57 (n = 9)7.72 ± 5.88 (n = 23)8.13 ± 5.11 (n = 8)0.087Height (SDS)0.11 ± 1.16 (n = 7)-2.17 ± 0.69 (n = 7)-0.91 ± 1.14 (n = 17)-0.75 ± 2.26 (n = 6)0.025Weight (SDS)0.29 ± 1.65 (n = 10)-2.23 ± 1.54 (n = 8)-0.55 ± 1.17 (n = 21)-0.86 ± 2.09 (n = 7)0.009Penile length (cm)2.75 ± 1.11 (n = 10)1.81 ± 0.86 (n = 8)2.45 ± 1.02(n = 20)2.53 ± 1.02(n = 7)0.275Testicular volume (mL)1.08 ± 0.53 (n = 10)0.73 ± 0.30 (n = 8)1.18 ± 0.74(n = 18)1.60 ± 0.55(n = 5)0.088Clinical features Growth retardation (height < -2 SD)0.00% (0/7)57.14% (4/7)17.65% (3/17)33.33% (2/6)0.063 Micropenis90.91% (10/11)100.00% (9/9)100.00% (23/23)100.00% (8/8)0.549 Cryptorchidism54.55% (6/11)44.44% (4/9)47.83% (11/23)50.00% (4/8)1.000 Secondary sexual characteristics0.00% (0/11)0.00% (0/9)0.00% (0/23)0.00% (0/8)NA Impaired olfactory function87.50% (7/8)50.00% (1/2)15.38% (2/13)16.67% (1/6)0.002 Facial anomaly0.00% (0/11)55.56% (5/9)0.00% (0/23)12.50% (1/8)0.000 Ocular abnormality0.00% (0/11)44.44% (4/9)4.35% (1/23)0.00% (0/8)0.007 Hearing loss0.00% (0/11)44.44% (4/9)0.00% (0/23)12.50% (1/8)0.001 Laryngeal problems0.00% (0/11)22.22% (2/9)0.00% (0/23)0.00% (0/8)0.050 Congenital heart disease0.00% (0/11)55.56% (5/9)4.35% (1/23)0.00% (0/8)0.001 Gastroesophageal reflux0.00% (0/11)11.11% (1/9)0.00% (0/23)0.00% (0/8)0.333 Unilateral absence of kidney18.18% (2/11)0.00% (0/9)0.00% (0/23)0.00% (0/8)0.093 Skeletal issues0.00% (0/11)11.11% (1/9)0.00% (0/23)12.50% (1/8)0.150 Developmental delay0.00% (0/11)33.33% (3/9)4.35% (1/23)25.00% (2/8)0.032 Weakness0.00% (0/11)11.11% (1/9)0.00% (0/23)0.00% (0/8)0.333 Enuresis0.00% (0/11)11.11% (1/9)0.00% (0/23)0.00% (0/8)0.333Imaging findings Delayed bone age50.00% (3/6)100.00% (2/2)66.67% (8/12)0.00% (0/1)0.454 Abnormal ultrasound of sexual glands85.71% (6/7)100.00% (7/7)88.24% (15/17)71.43% (5/7)0.708 Abnormal pituitary MRI40.00% (2/5)60.00% (3/5)40.00% (2/5)50.00% (1/2)1.000Gonadal hormones^c^ Basal FSH (IU/L)0.41 ± 0.22 (n = 11)1.55 ± 1.23 (n = 9)0.65 ± 0.49 (n = 23)0.91 ± 0.81 (n = 8)0.012 Basal LH (IU/L)0.08 ± 0.02 (n = 11)0.41 ± 0.54 (n = 9)0.12 ± 0.14 (n = 23)0.09 ± 0.06 (n = 8)0.191 Basal T (nmol/L)0.67 ± 0.66 (n = 11)0.39 ± 0.31 (n = 9)0.41 ± 0.26 (n = 23)0.36 ± 0.21 (n = 8)0.613 Basal AMH (ng/mL)7.61 ± 7.70 (n = 7)50.92 ± 68.67 (n = 8)29.11 ± 19.54 (n = 20)47.73 ± 15.36 (n = 6)0.003 Basal INHB (pg/mL)26.40 ± 12.30 (n = 6)64.91 ± 75.55 (n = 7)33.38 ± 35.55 (n = 18)42.35 ± 18.65 (n = 5)0.342 Basal LH < 1.0 IU/L100.00% (11/11)88.89% (8/9)100.00% (23/23)100.00% (8/8)0.333 Basal T < 3.5 nmol/L100.00% (11/11)100.00% (9/9)100.00% (23/23)100.00% (8/8)NA Low basal AMH71.43% (5/7)62.50% (5/8)40.00% (8/20)33.33% (2/6)0.395 Low basal INHB50.00% (3/6)14.29% (1/7)55.56% (10/18)20.00% (1/5)0.221 Peak FSH after GnRH stimulation (IU/L)1.23 ± 1.20 (n = 5)8.95 (n = 1)2.38 ± 1.05 (n = 4)3.98 (n = 1)0.177^b^ Peak LH after GnRH stimulation (IU/L)0.39 ± 0.27 (n = 5)7.63 (n = 1)1.09 ± 0.82 (n = 4)3.87 (n = 1)0.107^b^ LH/FSH after GnRH stimulation0.39 ± 0.15 (n = 5)0.85 (n = 1)0.51 ± 0.48 (n = 4)0.97 (n = 1)0.556^b^ T after HCG stimulation (nmol/L)0.58 ± 0.27 (n = 4)5.57 ± 5.38 (n = 4)1.07 ± 1.19 (n = 12)3.00 ± 2.45 (n = 5)0.103 AMH after HCG stimulation (ng/mL)29.22 ± 20.68 (n = 4)29.78 ± 26.76 (n = 4)25.25 ± 19.89 (n = 11)48.28 ± 12.87 (n = 4)0.689 INHB after HCG stimulation (pg/mL)35.69 ± 10.82 (n = 4)38.51 ± 21.04 (n = 4)35.30 ± 23.78 (n = 10)76.00 ± 18.21 (n = 3)0.052SDS, standard deviation score; FSH, follicle-stimulating hormone; LH, luteinizing hormone; T, testosterone; AMH, anti-mullerian hormone; INHB, inhibin B; GnRH, gonadotropin-releasing hormone^a^The p value comparing means among 4 groups was calculated using One-Way ANOVA or Kruskal-Wallis H test, except for those marked with an asterisk. The p value of cross-table was calculated using Fisher’s exact test^b^The p value comparing means between ANOS1 and FGFR1 group was calculated using student’s t test or Mann-Whitney U test^c^Those gonadal hormones below the lower detectable limit were valued with the lower detectable limit
Treatment and follow-up
Among the 51 patients, 13 were lost to follow-up, 4 were newly diagnosed without treatment, whereas 34 maintained follow-up and medication. All 34 patients were treated with HCG, including 28 (28/34, 82.35%) with HCG monotherapy and 6 (6/34, 17.65%) receiving HCG and HMG combination therapy. Seven of them (7/34, 20.59%) were further supplemented with TU, and 3 (3/34, 8.82%), P42, P50 and P51, were transitioned to pulsatile GnRH treatment.
Clinical follow-up lasted from 0.17 to 6.92 years with a mean time of 2.75 ± 1.82 years. After treatment, the levels of FSH, LH and testosterone elevated markedly, whereas AMH and INHB remained unchanged. Penile growth occurred in 21 patients (21/34, 61.76%), and testicular enlargement presented in 11 (11/34, 32.35%). Statistical analysis confirmed the significant increase of mean penile length, but the mean testicular volume was not significantly changed. Notably, 3 unrelated cases, P25, P48 and P50, who were administrated with HCG monotherapy, a combination therapy of HCG, HMG and TU, and GnRH therapy transitioned from HCG along with TU, respectively, showed optimal drug responses and gained the best benefits (Table 4, Table S5, Table S6 and Fig. 1).
Table 4. Comparison of clinical and hormonal parameters of 34 male IHH patients before and after treatmentPatientTreatmentFollow-up time (years)Penile length (cm)Testicular volume (mL)FSH (IU/L)^a^LH (IU/L)^a^T (nmol/L)^a^BeforeAfterBeforeAfterBeforeAfterBeforeAfterBeforeAfterP1HCG1.5012.5UnmeasurableUnmeasurable1.763.050.120.240.3810.45P2HCG6.9214UnmeasurableLeft: 1Right: 0.5< 0.31.15< 0.070.091.229.7P4HCG + HMG3.16130.51< 0.30.33< 0.070.090.389.71P5HCG6.83Unknown2.2Unmeasurable0.50.681.09< 0.07< 0.070.920.57P6HCG1.3312.50.50.54.323.10.730.3< 0.2424.37P7HCG1.671.42Unmeasurable0.50.50.86< 0.07< 0.07< 0.2412.59P9HCG0.841.81.80.50.50.76ND< 0.07ND< 0.2412.29P11HCG + HMG5.831.31.70.50.50.4ND< 0.07ND< 0.249.06P12HCG0.831.41.50.50.51.011.240.08< 0.07< 0.244.51P15HCG5.2512Left: UnmeasurableRight: 10.5< 0.3ND< 0.07ND0.353.51P18HCG2.592.32.60.50.5< 0.3ND0.14ND< 0.2420.33P19HCG0.593.53.5220.670.550.090.22< 0.241.59P20HCG0.332.52.50.50.50.44ND0.13ND< 0.2411.11P22HCG + TU2.4222.50.50.5< 0.31.2< 0.070.11< 0.243.24P24HCG5.002.53UnmeasurableLeft: UnmeasurableRight: 1< 0.3< 0.3< 0.070.18< 0.240.29P25HCG5.6735.6112< 0.33.2< 0.071.320.85.23P27HCG1.172.83.5111.8ND0.07ND< 0.2413.77P28HCG + TU3.173.3412.5< 0.31.140.10.25< 0.241.61P30HCG + HMG2.922.54.50.51< 0.30.76< 0.070.17< 0.2411.93P31HCG + HMG + TU2.582.52.522< 0.30.6< 0.070.130.364.28P33HCG3.753311.5< 0.3< 0.3< 0.07< 0.07< 0.24< 0.24P34HCG1.25441.520.480.650.110.190.345.21P36HCG1.671.53UnmeasurableUnmeasurable< 0.30.54< 0.070.110.831.01P38HCG3.503.54.5141.261.180.680.520.262.77P40HCG4.002211< 0.3< 0.3< 0.07< 0.070.461.39P42HCG transitioning to GnRH1.175523< 0.3< 0.3< 0.070.10.490.38P43HCG2.6724.5131.911.020.270.590.770.77P45HCG1.75UnknownUnknownLeft: 1Right: 0.5Left: 1Right: 0.5< 0.30.35< 0.07< 0.071.091.82P46HCG + HMG + TU3.333.5423< 0.30.980.240.380.442.66P47HCG0.173322< 0.3ND< 0.07ND0.522.35P48HCG + HMG + TU2.754636< 0.30.780.240.450.5726.85P49HCG1.0033110.690.98< 0.070.20.451.33P50HCG + TU transitioning to GnRH3.343616< 0.312.64< 0.079.121.5211.28P51HCG + TU transitioning to GnRH2.6744110.885.02< 0.073.562.225.5Mean ± SD2.75 ± 1.822.48 ± 1.073.32 ± 1.231.10 ± 0.651.95 ± 2.360.68 ± 0.791.62 ± 2.470.13 ± 0.150.69 ± 1.820.52 ± 0.446.87 ± 6.91p value^a^-0.0050.3160.0010.0010.000FSH, follicle-stimulating hormone; LH, luteinizing hormone; T, testosterone; HCG, human chorionic gonadotropin; HMG, human menopausal gonadotropin; GnRH, gonadotropin-releasing hormone; TU, testosterone undecanoate; ND, not done^a^The p value comparing means between before and after treatment group was calculated using student’s t test or Mann-Whitney U test^b^Those gonadal hormones below the lower detectable limit were valued with the lower detectable limit
Fig. 1. Comparison of clinical and hormonal parameters of 34 male IHH patients before and after treatment. A Penile length. B Testicular volume. C Follicle-stimulating hormone. D Luteinizing hormone. E Testosterone. F Anti-mullerian hormone. G Inhibin B. Results are presented as mean ± SD; ns, not significant; ** p < 0.01; ***p < 0.0001
Discussion
IHH is a set of rare diseases characterized by abnormal sexual development with clinical heterogeneity and genotypic complexity. To describe clinical and genetic features of male IHH patients in southern China and evaluate therapeutic effects of current treatments, we enrolled this cohort consisting of 51 male IHH patients.
Consistent with prior reports, our patients manifested typical clinical symptom of micropenis and absence of secondary sexual characteristics, and characteristic biochemical features of low testosterone along with low LH [3, 7]. However, the mean age at diagnosis of 7.84 ± 5.89 years in this study was much younger than previous studies [1, 2, 7, 8, 12, 18–22], probably due to the incorporation of genetic findings into case definition. IHH patients are usually not diagnosed until late adolescence or early adulthood based on clinical and biochemical manifestations. The wide use of genetic testing, especially NGS, accelerates the molecular diagnosis once the clinical diagnosis is made or suspected, suggesting that a combination of clinical, biochemical and genetic criteria will benefit early diagnosis.
In previous studies, variants in the FGFR1, ANOS1, PROKR2, and CHD7 gene account for most male IHH patients [1, 2, 4, 12, 23] (Table S7). In our study, the most frequent causative gene was FGFR1 (23/51, 45.10%), followed by ANOS1 (11/51, 21.57%) and CHD7 (9/51, 17.65%), revealing a similar molecular composition of male IHH between southern China and other countries or other regions of China. Twenty-three novel variants were identified in FGFR1, ANOS1 and CHD7 gene, which expands the mutational spectrum of IHH. Moreover, 8 patients were influenced by a paternal or maternal deleterious variant in the CHD7, PROKR2, FGFR1 and HS6ST1 gene, respectively. Although these genes can cause autosomal dominant IHH, the carrier parents were phenotypically normal and preserved fertility because of incomplete penetrance or highly phenotypic variability within families and among patients.
Age-phenotype and genotype-phenotype correlations were revealed in this study. The penile length, testicular volume and basal testosterone increased with age significantly, showing that the penis, testes and testosterone secretion could increase during puberty age, though they lacked obvious sexual development. ANOS1 and CHD7 patients rather than FGFR1 patients had distinctive features. Specifically, the family history, impaired olfactory function and much lower basal AMH were hallmarks for ANOS1 patients, whereas CHARGE phenotypes and higher basal FSH and LH could help to recognize CHD7 patients at younger age.
As basal AMH was much lower in patients with ANOS1 variants than other molecular causes, a differentially diagnostic value of AMH was prominent. Additionally, low basal AMH and INHB were displayed in 48.79% (20/41) and 41.67% (15/36) of our patients respectively, indicating their potential as diagnostic biomarkers for IHH, which fits well with previous studies [24–26].
After diagnosis, 34 patients were treated with HCG alone or conjugated with HMG. Among them, oral TU was added to the therapeutic schedule of 7 patients, while 3 patients were transitioned to pulsatile GnRH treatment. The treatments lasting 2.75 ± 1.82 years were effective as they markedly increased the penile length, and the levels of FSH, LH and testosterone, and yielded an increase of penile length in 21 patients (61.76%) and an enlargement of testicular volume in 11 patients (32.35%). No significant difference was shown among different therapeutic strategies. Three patients responded optimally to different strategies, highlighting the need of personalized approaches.
In fact, limitations exist in this retrospective study. As we know, GnRH stimulation test is crucial to the diagnosis of IHH. However, only 21.57% (11/51) of our patients were willing to receive GnRH stimulation test. Future work should focus on strategies to improve patient participation in GnRH stimulation test or to develop alternative diagnostic methods.
In this study, most cases were pediatric patients whose clinical phenotypes and gonadal hormone levels overlapped with those of both children with other disorders of sexual development and even normal controls. A definitive IHH diagnosis could not be made base on clinical signs and gonadal hormones without molecular evidence. Therefore, we incorporated genetic etiology as the fourth diagnostic criterion for IHH. Future work could further investigate the overall diagnostic yield of genetic testing when molecularly negative cases eventually meet definitive diagnostic criteria.
Conclusions
Our study reports 51 male IHH patients from southern China, describes their phenotypic and genotypic characteristics, and shares our experience with the follow-up and therapeutic effects, which enriches the patient resources and clinical data. Our study reveals FGFR1, ANOS1 and CHD7 as the most common causes of IHH in southern China, and finds 23 novel variants to extend the mutational spectrum of IHH. Our study indicates that a combination of clinical, biochemical and genetic criteria will facilitate early diagnosis. Our study also suggests that the family history, impaired olfactory function, CHARGE features, and basal AMH, FSH and LH have diagnostic values in distinguishing different molecular bases.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3
Supplementary Material 4
Supplementary Material 5
Supplementary Material 6
Supplementary Material 7
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Balasubramanian R, Crowley WF Jr., Isolated Gonadotropin-Releasing Hormone (Gn RH) Deficiency. 2007 May 23 [updated 2022 May 12]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024.20301509 · pubmed ↗