F8 Variants and Inhibitor Development in a Multiethnic Cohort of Nonsevere Haemophilia A
Ming Y. Lim, Kristy Lee, Jill M. Johnsen, Nigel S. Key

TL;DR
This study identifies F8 gene variants linked to inhibitor development in a diverse group of people with mild hemophilia A, showing these variants are common and not tied to race.
Contribution
The study expands the known F8 variants associated with inhibitor development in a multiethnic cohort, offering actionable insights for clinical management.
Findings
Inhibitors developed in 7.9% of the NSHA cohort, linked to 70 F8 variants.
Most inhibitor-associated F8 variants (95.7%) were missense mutations.
Race or ethnicity was not associated with inhibitor development in this multiethnic cohort.
Abstract
Neutralising antibodies (inhibitors) against factor VIII can result in severe bleeding in persons with nonsevere haemophilia A (NSHA). The INSIGHT study of 1112 persons with NSHA in a predominantly White population identified 19 different F8 missense variants that were associated with inhibitor development. To describe the F8 variants and inhibitor development in persons with NSHA in a multiethnic cohort using the My Life, Our Future (MLOF) Research Repository and the American Thrombosis and Hemostasis Network dataset (ATHNdataset). The MLOF Research Repository and ATHNdataset were queried for demographic, clinical and genotyping data. A total of 1805 persons with NSHA with at least one reportable F8 variant and known inhibitor status were included in this study. Inhibitors were developed in 142 (7.9%) persons with NSHA. Inhibitor development occurred in seventy F8 variants, of which…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
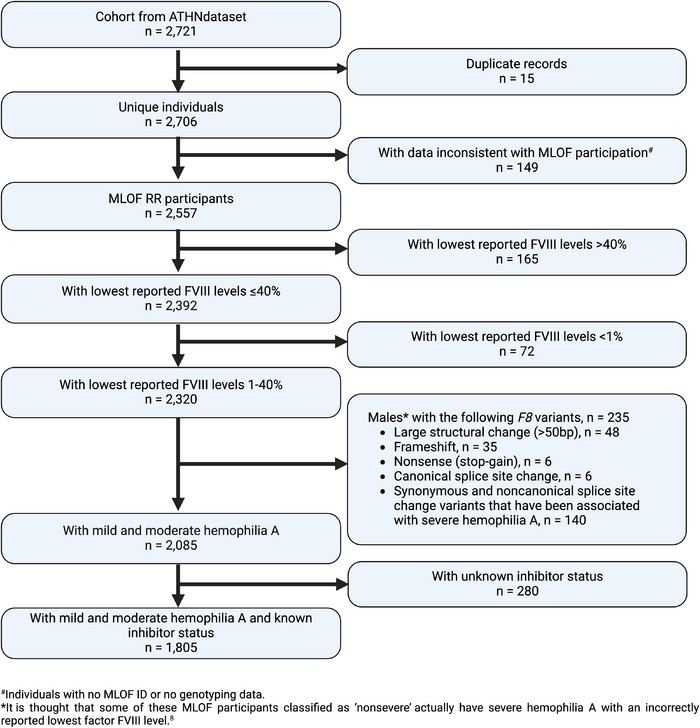 FIGURE 1
FIGURE 1| Overall ( | No inhibitor ( | Inhibitor ( | ||||
|---|---|---|---|---|---|---|
| Demographics |
| % |
| % |
| % |
|
| ||||||
| <2 years | 13 | 0.7 | 12 | 0.7 | 1 | 0.7 |
| 2 to 10 | 314 | 17.4 | 297 | 17.9 | 17 | 12.0 |
| 11 to 19 | 421 | 23.3 | 394 | 23.7 | 27 | 19.0 |
| 20 to 44 | 572 | 31.7 | 525 | 31.6 | 47 | 33.1 |
| 45 to 64 | 309 | 17.1 | 279 | 16.8 | 30 | 21.1 |
| 65+ | 157 | 8.7 | 141 | 8.5 | 16 | 11.3 |
| Died | 19 | 1.1 | 15 | 0.9 | 4 | 2.8 |
|
| ||||||
| 1921 to 1940 | 39 | 2.2 | 35 | 2.1 | 4 | 2.8 |
| 1941 to 1960 | 255 | 14.1 | 230 | 13.8 | 25 | 17.6 |
| 1961 to 1980 | 319 | 17.7 | 291 | 17.5 | 28 | 19.7 |
| 1981 to 2000 | 570 | 31.6 | 519 | 31.2 | 51 | 35.9 |
| 2001 to 2017 | 622 | 34.5 | 588 | 35.4 | 34 | 23.9 |
|
| ||||||
| Male | 1593 | 88.3 | 1452 | 87.3 | 141 | 99.3 |
| Female | 212 | 11.7 | 211 | 12.7 | 1 | 0.7 |
|
| ||||||
| White | 1541 | 85.4 | 1417 | 85.2 | 124 | 87.3 |
| Black | 121 | 6.7 | 110 | 6.6 | 11 | 7.7 |
| Asian | 38 | 2.1 | 36 | 2.2 | 2 | 1.4 |
| Other | 56 | 3.1 | 55 | 3.3 | 1 | 0.7 |
| Unknown | 49 | 2.7 | 45 | 2.7 | 4 | 2.8 |
|
| ||||||
| Hispanic | 270 | 15.0 | 251 | 15.1 | 19 | 13.4 |
| Non‐Hispanic | 1519 | 84.2 | 1397 | 84.0 | 122 | 85.9 |
| Unknown | 16 | 0.9 | 15 | 0.9 | 1 | 0.7 |
|
| ||||||
| Mild | 1194 | 66.1 | 1136 | 68.3 | 58 | 40.8 |
| Moderate | 611 | 33.9 | 527 | 31.7 | 84 | 59.2 |
|
| ||||||
| No | 1776 | 98.4 | 1638 | 98.5 | 138 | 97.2 |
| Yes | 29 | 1.6 | 25 | 1.5 | 4 | 2.8 |
|
| ||||||
| No | 1651 | 91.5 | 1528 | 91.9 | 123 | 86.6 |
| Yes | 154 | 8.5 | 135 | 8.1 | 19 | 13.4 |
|
| ||||||
| No | 1764 | 97.7 | 1628 | 97.9 | 136 | 95.8 |
| Yes | 41 | 2.3 | 35 | 2.1 | 6 | 4.2 |
| Variant Impact (Variant Type) | HGVS cDNA | HGVS Protein |
No. of persons with variant in overall cohort,
|
No. of persons with inhibitors,
| No. of persons with clinically relevant FVIII inhibitors, | Notes |
|---|---|---|---|---|---|---|
| Missense | c.80G>A | p.Gly27Asp | 1 (0.1) | 1 (100.0) | ||
| Missense | c.311T>A | p.Val104Asp | 9 (0.5) | 1 (11.1) | 1 | |
| Missense | c.344T>C | p.Val115Ala | 5 (0.3) | 1 (20.0) | ||
| Missense | c.403G>C | p.Asp135His | 1 (0.1) | 1 (100.0) | ||
| Missense | c.490G>A | p.Gly164Ser | 1 (0.1) | 1 (100.0) | 1 | |
| Missense | c.544G>T | p.Asp182Tyr | 8 (0.4) | 1 (12.5) | ||
| Missense | c.818A>G | p.Tyr273Cys | 1 (0.1) | 1 (100.0) | ||
| Missense | c.871G>A | p.Glu291Lys | 20 (1.1) | 1 (5.0) | Reported in 17 hemizygous males and 3 heterozygous females | |
| Missense | c.878A>G | p.His293Arg | 1 (0.1) | 1 (100.0) | 1 | |
| Missense | c.935T>C | p.Phe312Ser | 46 (2.5) | 3 (6.5) | 2 | Reported in 44 hemizygous males and 2 heterozygous females |
| Missense | c.1015A>G | p.Met339Val | 5 (0.3) | 1 (20.0) | ||
| Missense | c.1172G>A | p.Arg391His | 23 (1.3) | 2 (8.7) | 1 | Reported in 21 hemizygous males and 2 heterozygous females |
| Missense | c.1312A>T | p.Ile438Phe | 3 (0.2) | 1 (33.3) | ||
| Missense | c.1316G>T | p.Gly439Val | 10 (0.6) | 1 (10.0) | 1 | |
| Missense | c.1397G>A | p.Gly466Glu | 1 (0.1) | 1 (100.0) | ||
| Missense | c.1475A>G | p.Tyr492Cys | 3 (0.2) | 1 (33.3) | ||
| Missense | c.1478A>C | p.Asn493Thr | 1 (0.1) | 1 (100.0) | ||
| Missense | c.1594T>G | p.Trp532Gly | 1 (0.1) | 1 (100.0) | 1 | |
| Missense | c.1636C>T | p.Arg546Trp | 37 (2.0) | 1 (2.7) | Reported in 33 hemizygous males and 4 heterozygous females | |
| Missense | c.1648C>T | p.Arg550Cys | 31 (1.7) | 2 (6.5) | 1 | |
| Missense | c.1649G>A | p.Arg550His | 14 (0.8) | 3 (21.4) | 1 | Reported in 13 hemizygous males and 1 heterozygous female |
| Missense | c.1660A>G | p.Ser554Gly | 58 (3.2) | 4 (6.9) | Reported in 57 hemizygous males and 1 heterozygous female | |
| Missense | c.1813T>C | p.Tyr605His | 1 (0.1) | 1 (100.0) | ||
| Missense | c.1834C>T | p.Arg612Cys | 103 (5.7) | 12 (11.7) | 5 | Reported in 102 hemizygous males and 1 heterozygous female. The female has an additional causative variant (c.[5954delG]) |
| Missense | c.2053G>A | p.Asp685Asn | 1 (0.1) | 1 (100.0) | ||
| Missense | c.2087C>T | p.Thr696Ile | 7 (0.4) | 1 (14.3) | ||
| Missense | c.2101A>G | p.Met701Val | 2 (0.1) | 1 (50.0) | ||
| Missense | c.2167G>A | p.Ala723Thr | 116 (6.4) | 9 (7.8) | Reported in 110 hemizygous males and 6 heterozygous females. One female has an additional causative variant (c.[6066C>G]) | |
| Missense | c.5096A>C | p.Tyr1699Ser | 4 (0.2) | 3 (75.0) | ||
| Missense | c.5096A>T | p.Tyr1699Phe | 43 (2.4) | 3 (7.0) | Reported in 40 hemizygous males and 3 heterozygous females | |
| Missense | c.5122C>T | p.Arg1708Cys | 7 (0.4) | 2 (28.6) | ||
| Missense | c.5399G>A | p.Arg1800His | 28 (1.6) | 1 (3.6) | Reported in 27 hemizygous males and 1 heterozygous female | |
| Missense | c.5721C>G | p.Ser1907Arg | 1 (0.1) | 1 (100.0) | ||
| Missense | c.5726A>G | p.Tyr1909Cys | 1 (0.1) | 1 (100.0) | ||
| Missense | c.5822A>G | p.Asn1941Ser | 46 (2.5) | 6 (13.0) | 3 | Reported in 43 hemizygous males and 3 heterozygous females |
| Missense | c.5843T>C | p.Leu1948Pro | 8 (0.4) | 2 (25.0) | 1 | |
| Missense | c.5879G>A | p.Arg1960Gln | 16 (0.9) | 5 (31.3) | ||
| Missense | c.6016G>A | p.Glu2006Lys | 5 (0.3) | 1 (20.0) | ||
| Missense | c.6046C>T | p.Arg2016Trp | 20 (1.1) | 1 (5.0) | Reported in 19 hemizygous males and 1 heterozygous female | |
| Missense | c.6113A>G | p.Asn2038Ser | 10 (0.6) | 2 (20.0) | 1 | Reported in 9 hemizygous males and 1 heterozygous female |
| Missense | c.6206T>C | p.Leu2069Pro | 1 (0.1) | 1 (100.0) | ||
| Missense | c.6265T>C | p.Trp2089Arg | 1 (0.1) | 1 (100.0) | ||
| Missense | c.6267G>T | p.Trp2089Cys | 1 (0.1) | 1 (100.0) | ||
| Missense | c.6277G>C | p.Asp2093His | 3 (0.2) | 1 (33.3) | 1 | |
| Missense | c.6316C>G | p.Gln2106Glu | 5 (0.3) | 1 (20.0) | 1 | |
| Missense | c.6320G>A | p.Gly2107Asp | 2 (0.1) | 1 (50.0) | ||
| Missense | c.6371A>G | p.Tyr2124Cys | 15 (0.8) | 4 (26.7) | 2 | Reported in 14 hemizygous males and 1 heterozygous female |
| Missense | c.6393G>T | p.Trp2131Cys | 1 (0.1) | 1 (100.0) | ||
| Missense | c.6443A>G | p.Asn2148Ser | 8 (0.4) | 1 (12.5) | ||
| Missense | c.6469A>G | p.Asn2157Asp | 4 (0.2) | 2 (50.0) | ||
| Missense | c.6506G>A | p.Arg2169His | 85 (4.7) | 21 (24.7) | 7 | Reported in 82 hemizygous males and 3 heterozygous females |
| Missense | c.6518C>A | p.Thr2173Asn | 3 (0.2) | 1 (33.3) | ||
| Missense | c.6533G>A | p.Arg2178His | 7 (0.4) | 1 (14.3) | ||
| Missense | c.6533G>T | p.Arg2178Leu | 15 (0.8) | 1 (6.7) | Reported in 14 hemizygous males and 1heterozygous female | |
| Missense | c.6544C>T | p.Arg2182Cys | 6 (0.3) | 1 (16.7) | Reported in 2 hemizygous males and 4 heterozygous females | |
| Missense | c.6551A>C | p.Glu2184Ala | 3 (0.2) | 1 (33.3) | ||
| Missense | c.6658G>C | p.Ala2220Pro | 14 (0.8) | 1 (7.1) | Reported in 13 hemizygous males and 1 heterozygous female | |
| Missense | c.6679G>A | p.Ala2227Thr | 8 (0.4) | 2 (25.0) | ||
| Missense | c.6683G>A | p.Arg2228Gln | 3 (0.2) | 1 (33.3) | ||
| Missense | c.6715A>G | p.Arg2239Gly | 3 (0.2) | 1 (33.3) | ||
| Missense | c.6752T>C | p.Val2251Ala | 3 (0.2) | 1 (33.3) | ||
| Missense | c.6795G>T | p.Gln2265His | 1 (0.1) | 1 (100.0) | ||
| Missense | c.6845C>T | p.Ser2282Phe | 1 (0.1) | 1 (100.0) | ||
| Missense | c.6967C>G | p.Arg2323Gly | 8 (0.4) | 1 (12.5) | ||
| Missense | c.6967C>T | p.Arg2323Cys | 9 (0.5) | 1 (11.1) | Reported in 8 hemizygous males and 1 heterozygous female | |
| Missense | c.6968G>C | p.Arg2323Pro | 5 (0.3) | 1 (20.0) | 1 | |
| Missense | c.6977G>A | p.Arg2326Gln | 18 (1.0) | 3 (16.7) | ||
| Splice site change | c.1903+5G>T | 1 (0.1) | 1 (100.0) | |||
| Splice site change | c.5219+3A>G | 10 (0.6) | 2 (20.0) | Reported in 8 hemizygous males and 2 heterozygous females | ||
| Large structural change (>50 bp) | c.[6429+?_6430‐?inv] | 63 (3.5) | 1 | Reported in 63 heterozygous females, for which 3 females had additional causative variant (c.[1‐?_6429+?del]; c.[6430‐?_6900+?dup]; c.[1569G>T]) | ||
|
| 1006 (55.7) | 142 | 32 |
- —Hemostasis and Thrombosis Research Society10.13039/100005881
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHemophilia Treatment and Research · Platelet Disorders and Treatments · Blood Coagulation and Thrombosis Mechanisms
Introduction
1
The development of neutralising antibodies, or inhibitors, against infused exogenous factor VIII (FVIII) represents a challenging complication in the treatment of haemophilia A. In persons with nonsevere haemophilia A (NSHA), inhibitor development is thought to be uncommon [1]. However, a landmark paper by Eckhardt et al. on behalf of the INSIGHT Study Group of 1112 persons with NSHA from 14 centres in Europe and Australia found that inhibitor development occurs throughout the lifetime in persons with NSHA, with a cumulative risk as high as 13.3% in individuals with >100 exposure days (ED) to FVIII [2]. Additionally, the INSIGHT study found that inhibitor development in persons with NSHA is associated with increased mortality [3] and thus deserves further evaluation.
The F8 variant is an important risk factor for inhibitor development [4, 5]. In individuals with severe haemophilia A (SHA), the risk for inhibitor development is increased by variants that are more likely to be null, such as inversions, complex structural variants, or nonsense (stop‐gain) variants of the F8 gene. In contrast, inhibitor development in NSHA is thought to be caused by F8 missense variants that likely cause conformational changes within immunogenic domains of F8 [6]. It has been demonstrated that among more than 150 different causative missense variants for NSHA, some relatively prevalent variants are associated with a high risk for inhibitor development [2, 6, 7]. The INSIGHT study found that among 214 different F8 missense variants, 19 variants were associated with inhibitor development [2]. In certain rare variants (p.Asp2074Gly and p.Trp2229Cys*)*, the risk for inhibitor development at 20 ED (21.2% and 41.7%, respectively) parallels that seen in persons with SHA. Although it is not entirely clear why these particular variants carry an increased risk for inhibitors, the INSIGHT study was a first step towards the identification of high‐risk persons with NSHA based on their F8 genotype.
Although the INSIGHT study was the largest study on persons with NSHA, it may not have captured other NSHA‐causing genotypes as it consisted of a predominantly White population (94.3%), with only 1.3% Black. Race and ethnicity are significantly associated with inhibitors in both haemophilia A and B [8] with the prevalence of FVIII inhibitors in the Black population about twice that of Whites [9]. Given this background, we set out to use the My Life, Our Future (MLOF) Research Repository to describe the F8 variants and inhibitor development in a multiethnic cohort of NSHA.
Methods
2
Subjects and Study Design
2.1
The MLOF Research Repository contains de‐identified biologic samples and data from participants who enrolled in the MLOF genotyping project at haemophilia treatment centres (HTCs) across the United States (US). Details on participant enrolment, clinical data submitted, and F8 variant screening and confirmation for the MLOF Research Repository can be found in Johnsen et al. [8]. All participants in MLOF also authorised for enrolment in the ATHNdataset. The ATHNdataset consists of a ‘limited dataset’ as defined under the Health Insurance Portability and Accountability Act to be free of protected health information. The ATHNdataset began enrolling alive persons with inherited bleeding disorders who have authorised sharing of their demographic and clinical information for research on 1 January 2010.
Data Collection and Definitions
2.2
MLOF eligibility included individuals with a clinical diagnosis of haemophilia A (for whom a reported FVIII level had to be <50%) and females at‐risk for or known to have a haemophilia A genotype (regardless of factor level). For this study, we included persons with NSHA who were categorised as moderate or mild haemophilia A using the lowest reported FVIII level of 1%–5% or >5%–40%, respectively [10, 11]. We excluded persons with FVIII levels of >40%. The datasets were queried on 31 December 2019 to extract the following: Demographics, haemophilia A severity, inhibitor status, F8 variants, current or past infection with hepatitis B and/or hepatitis C virus, and presence of HIV infection. Demographics included age, gender, self‐classified race (White, Black, Asian and Others), and ethnicity (Hispanic and non‐Hispanic). The lowest reported FVIII level submitted during enrolment to the MLOF Research Repository was used to assign haemophilia severity.
Male individuals with one reportable F8 variant consisting of either large structural change (>50 bp), frameshift, nonsense (stop‐gain), or canonical splicing (−1, −2, +1, or +2) variant for whom the lowest reported FVIII level was ≥1% were excluded from the study. It is thought that some of these MLOF participants classified as ‘nonsevere’ actually have SHA with an incorrectly reported lowest FVIII level [8]. For male individuals with one reportable F8 variant consisting of either synonymous or noncanonical splicing variants for whom the lowest reported FVIII level was ≥1%, both the European Association for Hemophilia and Allied Disorders (EAHAD) Coagulation Factor VIII Variant Database [12] and the Centers for Disease Control and Prevention Hemophilia A Mutation Project (CHAMP) [13] were reviewed and those whose F8 variants have been associated with SHA were also excluded. For male individuals with two reportable F8 variants (n = 13), the variant predicted to be the most impactful was used to determine exclusion. Eleven male individuals had two missense variants, while two male individuals had one missense variant and one non‐disease‐causing synonymous variant [8]. No males with two reportable F8 variants were excluded from this study. Female individuals who were heterozygous or compound heterozygous for any F8 variants were included if their lowest reported FVIII level was ≥1 IU/dL, indicating a diagnosis of mild or moderate haemophilia A.
Inhibitor testing was performed locally at the discretion of each HTC and recorded in the ATHNdataset. There is no mandatory requirement for inhibitor test reporting in the ATHNdataset. A person with NSHA was classified as positive for inhibitor if a detectable FVIII inhibitor of ≥0.6 Bethesda inhibitor assay unit per mL (BU/mL) was reported at any time in the ATHNdataset and/or if an inhibitor was indicated clinically in the ‘Inhibitor Status’ field (i.e., active, history of, inactive). Otherwise, a person with NSHA was considered not to have had an inhibitor if all reported inhibitor titres were <0.6 BU/mL or below the threshold of detection, and/or if the ‘Inhibitor Status’ field indicated no inhibitor (i.e., no history of, ruled out). If the relevant Inhibitor status field was uninformative (i.e., evaluation pending, unknown, or blank) and no historical inhibitor titre data were available, the subject with NSHA was classified as Unknown.
For persons with NSHA with a positive inhibitor status, we also determined if they had a clinically relevant FVIII inhibitor [14]. The definition of a clinically relevant FVIII inhibitor was adapted from the INSIGHT study [2] and has been used in prior studies using the ATHNdataset [15, 16]. Briefly, a clinically relevant FVIII inhibitor was defined as having at least 2 inhibitor titres of ≥1.0 BU/mL at two different time points. Although the INSIGHT study also included persons with inhibitor titres between 0.6 and 1.0 BU/mL who had a decrease in factor VIII plasma level to at least 50% of the baseline level, or a reduced half‐life after factor VIII administration of <6 h, these data were not consistently available in the ATHNdataset and were not used in this study.
Statistical Analysis
2.3
We used descriptive statistical analyses. Categorical variables were expressed as frequencies and percentage values. The association of inhibitor status with race (White, Black, and Asian), ethnicity (Hispanic, and non‐Hispanic) and viral infections (Hepatitis B, Hepatitis C, and HIV infections) were performed using χ^2^ tests of independence with a predetermined level of significance of 0.05. Individuals of ‘other’ or ‘unknown’ race and ethnicity were excluded due to insufficient data. All statistical analyses were performed using Stata MP v16 (College Station, TX, USA).
Results and Discussion
3
Patient Characteristics
3.1
A total of 1805 persons with NSHA with genotyping data and known inhibitor status were included in this study (Figure 1). The demographics and clinical characteristics of the NSHA cohort are shown in Table 1. Overall, the age distribution in the MLOF NSHA cohort was comparable with the NSHA data from the population of US HTCs from 2012 to 2022 [17].
Flow chart describing data cleaning to identify persons with nonsevere haemophilia A by lowest reported FVIII levels with genotyping data and known inhibitor status.
F8 variants and Inhibitor Development
3.2
Inhibitors developed in 142 (7.9%) persons with NSHA (males, n = 141; females, n = 1). Inhibitor development occurred in seventy F8 variants, of which the majority (n = 67, 95.7%) were missense variants (Table 2). These seventy *F8 *variants were identified in a total of 1006 (55.7%) persons with NSHA. The female individual with inhibitor had a baseline FVIII level of 30% and a heterozygous large structural change F8 variant (c.[6429+?_6430‐?inv]).
Seven F8 missense variants were reported in >40 persons with NSHA. Inhibitor development occurred in all seven F8 missense variants: p.Ala723Thr (9/116, 7.8%), p.Arg612Cys (12/103, 11.7%), p.Arg2169His (21/85, 24.7%), p.Ser554Gly (4/58, 6.9%), p.Asn1941Ser (6/46, 13.0%), p.Phe312Ser (3/46, 6.5%), and p.Tyr1699Phe (3/43, 7.0%).
Thirty‐two persons with NSHA, comprising 18 F8 missense variants, had a clinically relevant FVIII inhibitor (Table 2). The three most common F8 missense variants observed in those with inhibitors and those with clinically relevant FVIII inhibitors were c.1834C>T (p.Arg612Cys), c.5822A>G (p.Asn1941Ser), and c.6506G>A (p.Arg2169His).
Association of Inhibitor Development and Race, Ethnicity, or Viral Infection
3.3
In persons with NSHA, a χ ^2^ test of independence of the three racial groups (White, Black and Asian) and inhibitor status found no association between race and inhibitors (Table S1). Similarly, there was no association between ethnicity and inhibitors. Inhibitors were associated with infection with hepatitis C (χ^2^ = 4.6426, p‐value = 0.031188), but not with hepatitis B and HIV infection.
Discussion
4
We describe the largest cohort of persons with NSHA and inhibitor development using the MLOF Research Repository and a uniform data collection tool – the ATHNdataset. This study identified 70 F8 variants where inhibitor development occurred in a multiethnic cohort of NSHA. Of these 70 variants, six (p.Arg550Cys, p.Arg612Cys, p.Arg2169His, p.Arg2016Trp, p.Tyr2124Cys, p.Val2251Ala) were also identified in the INSIGHT study [2]. Overall, the MLOF cohort identified an additional 64 F8 variants where inhibitor development occurred in our multiethnic cohort. Knowledge of inhibitor development occurring in these F8 variants could help identify at‐risk individuals with NSHA based on their F8 variant. Specifically, three of the most common F8 variants that were seen in our study, namely p.Arg612Cys, p.Arg2169His, and p.Asn1941Ser had a rate of inhibitor development of >10%. Two of these, p.Arg612Cys and p.Arg2169His, were observed as well in the INSIGHT study and had a cumulative inhibitor risk of 18.3% and 12.2%, respectively, at 50 EDs [2] – a risk that approaches that of SHA. Identification of these at‐risk individuals can inform both physicians and persons with NSHA to adopt measures to reduce the risk of inhibitor development. These measures include the use of desmopressin or avoiding intensive courses of treatment with FVIII concentrates where possible.
In our study of NSHA with inhibitors, race or ethnicity was not associated with inhibitor development. This is in contrast to prior studies, including the MLOF parent study, which found that Black and Asian individuals, and those of Hispanic ethnicity, had significantly higher inhibitor rates than White individuals [8, 18, 19, 20, 21]. These prior studies all included SHA, which may account for the discrepant findings. It is also possible that the lack of association seen is due to insufficient power from the smaller number of NSHA individuals with inhibitors. Notably, this sub‐study of NSHA within the MLOF cohort had a lower prevalence of Black participants (6.7%) than the parent study (9.6%) [8], suggesting an under‐representation of minorities with NSHA in haemophilia clinical studies.
This study has several limitations. First, the classification of NSHA was based on the lowest recorded FVIII level at participating HTCs. This led to discrepancies in the classification of severity and was addressed by removing male individuals with null F8 variants or variants that have been associated with SHA. Second, inhibitor data entry in the ATHNdataset was not mandatory for participation in MLOF, which resulted in about ∼13% of subjects with unknown inhibitor status who were excluded from this analysis. These individuals were likely negative for inhibitors, as those with positive inhibitor status usually require more medical attention and would likely have had their data entered into the ATHNdataset. Lastly, the number of EDs to FVIII concentrates and detailed description on the circumstances surrounding inhibitor development were not available in the ATHNdataset. This limitation highlights a critical need for data collection on number of EDs and other risk factors for inhibitor development to develop a personalised inhibitor risk stratification model based on F8 variants.
Conclusion
5
The MLOF Research Repository offered a unique opportunity to uncover insights previously not possible, such as additional F8 variants where inhibitor development occurred in a multiethnic cohort. Additionally, race and ethnicity were not associated with inhibitor development in persons with NSHA and inhibitors. Identifying persons with NSHA at‐risk for inhibitor development is important for the clinical management of this population.
Author Contributions
Ming Y. Lim, Kristy Lee, Jill M. Johnsen, and Nigel S. Key participated in study methods design, data analysis, interpretation of results, and writing the manuscript.
Ethics Statement
As the data obtained for this study were deidentified, this study was considered nonhuman subject research by the University of Utah institutional review board.
Conflicts of Interest
M.Y.L. reports advisory board participation for Sanofi, Biomarin, Bayer, and Hema Biologics, and her institution has received research funding on her behalf from Sanofi. K.L. has no relevant disclosures. J.M.J. reports consulting for CSL Behring, Biomarin, Octapharma, and Takeda, and her institution has received research funding on her behalf from Octapharma. N.S.K. reports consulting for Pfizer, Centessa, and Novo Nordisk.
Supporting information
Supplemental Table 1: Association of inhibitor development and race, ethnicity, or viral infection.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1J. Wight and S. Paisley , “The Epidemiology of Inhibitors in Haemophilia A: A Systematic Review,” Haemophilia 9, no. 4 (2003): 418–435, 10.1046/j.1365-2516.2003.00780.x.12828678 · doi ↗ · pubmed ↗
- 2C. L. Eckhardt , A. S. van Velzen , M. Peters , et al., “Factor VIII gene (F 8) Mutation and Risk of Inhibitor Development in Nonsevere Hemophilia A,” Blood 122, no. 11 (2013): 1954–1962, 10.1182/blood-2013-02-483263.23926300 · doi ↗ · pubmed ↗
- 3C. L. Eckhardt , J. I. Loomans , A. S. van Velzen , et al., “Inhibitor Development and Mortality in Non‐Severe Hemophilia A,” Journal of Thrombosis and Haemostasis 13, no. 7 (2015): 1217–1225, 10.1111/jth.12990.25912309 · doi ↗ · pubmed ↗
- 4J. Boekhorst , G. R. Lari , and R. D'Oiron , “Factor VIII Genotype and Inhibitor Development in Patients With Haemophilia A: Highest Risk in Patients With Splice Site Mutations,” Haemophilia 14, no. 4 (2008): 729–735, 10.1111/j.1365-2516.2008.01694.x.18503540 · doi ↗ · pubmed ↗
- 5S. C. Gouw , H. M. van den Berg , J. Oldenburg , et al., “F 8 Gene Mutation Type and Inhibitor Development in Patients With Severe Hemophilia A: Systematic Review and Meta‐Analysis,” Blood 119, no. 12 (2012): 2922–2934, 10.1182/blood-2011-09-379453.22282501 · doi ↗ · pubmed ↗
- 6R. d'Oiron , S. W. Pipe , and M. Jacquemin , “Mild/Moderate Haemophilia A: New Insights Into Molecular Mechanisms and Inhibitor Development,” Haemophilia 14, no. 3 (2008): 138–146.18510534 10.1111/j.1365-2516.2008.01730.x · doi ↗ · pubmed ↗
- 7C. R. Hay , “Factor VIII Inhibitors in Mild and Moderate‐Severity Haemophilia A,” Haemophilia 4, no. 4 (1998): 558–563.9873794 10.1046/j.1365-2516.1998.440558.x · doi ↗ · pubmed ↗
- 8J. M. Johnsen , S. N. Fletcher , A. Dove , et al., “Results of Genetic Analysis of 11 341 Participants Enrolled in the My Life, Our Future Hemophilia Genotyping Initiative in the United States,” Journal of Thrombosis and Haemostasis 20, no. 9 (2022): 2022–2034, 10.1111/jth.15805.35770352 · doi ↗ · pubmed ↗