Response to anaplastic lymphoma kinase inhibitor in gastric cancer harboring DCTN1–ALK fusion: a case report and review
Huadi Wang, Liangkun You, Hong Pan, Xiaotong Qiu, Jin Sheng

TL;DR
A patient with gastric cancer responded to an ALK inhibitor after failing standard treatments, showing the importance of genomic profiling.
Contribution
First reported case of DCTN1–ALK fusion in gastric cancer successfully treated with alectinib.
Findings
Alectinib provided 11.5 months of progression-free survival in a gastric cancer patient with DCTN1–ALK fusion.
KRAS amplification detected via liquid biopsy led to rapid relapse after initial response to alectinib.
DCTN1–ALK fusion is an actionable driver gene in gastric cancer, responding to ALK inhibitors.
Abstract
Anaplastic lymphoma kinase (ALK) rearrangements are exceedingly rare in gastric cancer, and uncommon fusion types add to the difficulties of proper, precise treatment strategies. Although detected in non-small cell lung cancer (NSCLC), inflammatory myofibroblastic tumors (IMTs), and Spitz tumors, the DCTN1–ALK fusion has not previously been reported in gastric cancer. This report describes the first case of gastric adenocarcinoma harboring a DCTN1–ALK fusion that was successfully treated with the ALK-targeted agent alectinib after first- and second-line chemotherapy-based regimens had failed. Progression-free survival on alectinib was 11.5 months until KRAS amplification emerged on serial circulating tumor DNA analysis, leading to rapid systemic relapse. The other documented cases with DCTN1–ALK fusion treated with the first or second generation of ALK inhibitors indicated this rare…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
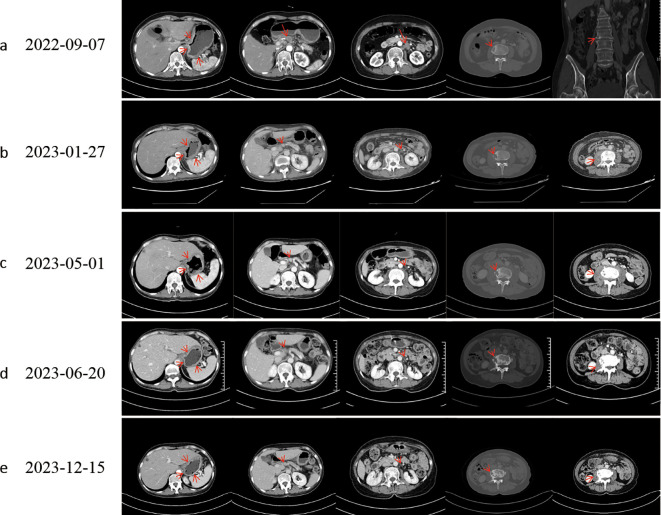 Figure 1
Figure 1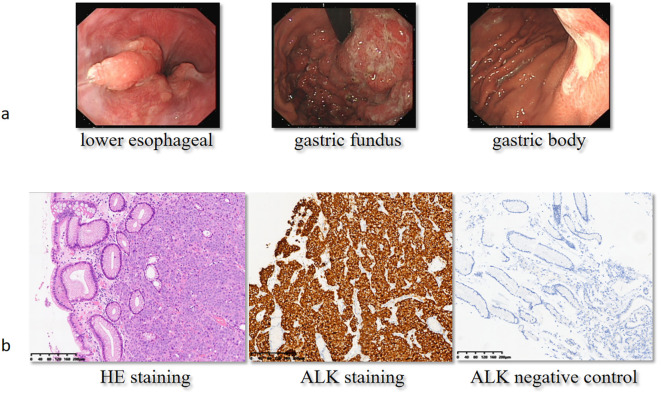 Figure 2
Figure 2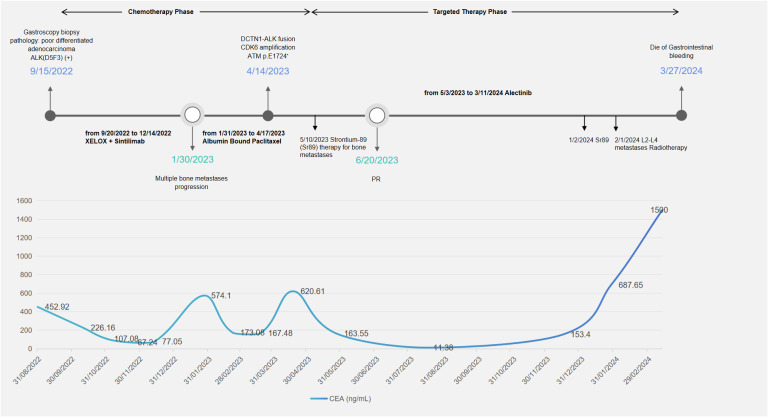 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLung Cancer Treatments and Mutations · Metastasis and carcinoma case studies · Gastrointestinal Tumor Research and Treatment
Introduction
Gastric cancer still poses a significant challenge in oncology due to its heterogeneity and unsatisfactory response to traditional chemotherapy. Recent advancements in cancer research have identified gastric cancer as subgroups based on epidemiologic (1), genomic (2), or molecular (3) classifications that may benefit from potential therapeutic targets. Among the various subtypes of gastrointestinal (GI) cancers, although very rare, anaplastic lymphoma kinase (ALK) rearrangements represent a particularly intriguing group, suggesting a potential for targeted therapy with ALK inhibitors (ALKi), offering a ray of hope for personalized treatment approaches.
The ALK gene encodes a receptor tyrosine kinase that is involved in cell growth, proliferation, and survival. Mutations in the ALK gene can lead to the aberrant activation of the ALK protein, resulting in uncontrolled cell growth and tumor formation. ALK rearrangements are extremely rare in GI cancers, with a frequency of <1% in colorectal cancer (4), 0.2% in pancreatic cancer (5), and 0.9% to 2.3% in gastric cancer (1, 6), but may offer personalized treatment strategies in selected patients. A Korean gastric cancer cohort (n = 455) consisted of 38 ALK Immunohistochemistry (IHC)-positive (34 of 1+, and 4 of 2+/3+) patients who were younger and more likely to have signet ring cells (1), associated with worse disease-free survival (DFS) and overall survival (OS) (1). Fortunately, ALKi, such as alectinib and lorlatinib (7), have demonstrated efficacy in inhibiting tumor growth and improving patient outcomes in ALK-mutant gastric cancer, offering a more effective and less toxic alternative to traditional chemotherapy.
The appearance of the DCTN1–ALK fusion gene in cancer is a rare genetic rearrangement event primarily associated with the occurrence and progression of tumors. Dynactin subunit 1 (DCTN1) is a protein related to the cytoskeleton, involved in the transport of materials within the cell. When DCTN1 undergoes a gene fusion with ALK, DCTN1 is suspected to promote the dimerization of ALK and lead to the abnormal activation of the ALK kinase by transphosphorylation, thereby promoting the proliferation and survival of tumor cells (8). To date, published case reports of DCTN1–ALK fusion solid tumors included non-small cell lung cancer (NSCLC) (9–11), inflammatory myofibroblastic tumors (IMTs) (12, 13), Spitz tumors (14), and some rare types of neoplasms.
Given the rarity of both ALK mutation and the fusion type, here, we report a case of a gastric cancer patient with a driver mutation of DCTN1–ALK fusion. Although the first and second lines of standard chemotherapy ended in progression of disease, subsequent targeted therapy improved the general health of the patient and prolonged the survival.
Case presentation
The patient, a 58-year-old woman, was admitted to the local hospital due to symptoms of cough, decline in appetite, and mild weight loss in August 2022. Laboratory tests revealed a significant increase in carcinoembryonic antigen (CEA) to 323.26 ng/mL and CA724 to 266.83 U/mL. The patient was then transferred to the Department of General Practice. Physical examinations did not show positive findings. A chest computed tomography (CT) scan showed enlarged mediastinal lymph nodes. An enhanced abdominal CT scan revealed thickening with enhancement of the lateral wall of the gastric cardia, fundus, and lesser curvature of the gastric body, indicating a T4a malignant tumor (Figure 1a). In addition, L3 vertebral metastasis was suspected. Multiple lymph node enlargements were observed in the perigastric, hilar, mesenteric, and retroperitoneal regions, consistent with metastasis (cT4aN3M1, stage IV). On September 8, 2022, the patient received a gastroduodenoscopy, and biopsies of the gastric angle, fundus, and lower esophagus were pathologically diagnosed as poorly differentiated carcinoma (Figure 2). Immunohistochemical analysis showed positive staining for cytokeratin (CK7), cytokeratin pan-cocktail (AE1/AE3), chromogranin A (focal positive), Ki-67 (40%), and ALK (D5F3) (Figure 2), and negative staining for S-100, thyroid transcription factor 1 (TTF1), hepatocyte, estrogen receptor, synaptophysin (Syn), CD56, CD3, CD20, GATA-3, NapsinA, SOX10, and human epidermal growth factor receptor 2 (HER2) (Supplementary Figure 1), as well as intact expression of mismatch repair proteins.
Computed tomography evaluation. Following effective therapy, the patient exhibited decreased gastric wall thickness, reduced osteolytic destruction of bone metastases, increased osteoblastic changes, and diminished perilesional soft-tissue components around osseous metastases. (a) Baseline. (b) Evaluation after progression of first-line chemotherapy. (c) Evaluation after progression of second-line chemotherapy; L3 vertebral metastasis showed progressive osteolytic destruction with an expanding perilesional soft-tissue mass. (d, e) The best response to targeted therapy was a partial response (PR).
Gastroduodenoscopy and immunohistochemical analysis at baseline. On September 8, 2022, the patient received a gastroduodenoscopy (a), and the pathologist reported poorly differentiated carcinoma with positive ALK staining (b). H&E, hematoxylin and eosin; ALK, anaplastic lymphoma kinase.
Standard chemotherapy phase
From September 20, 2022, to December 14, 2022, the patient received chemotherapy combined with immunotherapy for five cycles, consisting of capecitabine and oxaliplatin (XELOX) plus sintilimab. Meanwhile, a whole-body bone CT scan on November 22, 2022, showed multiple abnormal increases in bone metabolism at the seventh cervical vertebra, 10th and 11th thoracic vertebrae, third lumbar vertebra, right third anterior rib, sacrum, bilateral iliac bones, right ischium, and upper segment of the left femur, primarily indicating tumor bone metastasis. The patient received bisphosphonate therapy to retard osteolysis.
On January 27, 2023, due to elevated tumor markers (Figure 3) and increased soft tissue at the L3 metastatic lesion (Figure 1b), the disease was considered to be progressing. From January 31, 2023, to April 17, 2023, the patient received single-agent chemotherapy with albumin-bound paclitaxel as second-line treatment.
Timeline of treatment and CEA changes. During the chemotherapy phase, the patient received two lines of treatment: XELOX plus sintilimab and albumin-bound paclitaxel. The patient received strontium-89 therapy and localized radiotherapy for bone metastases. XELOX, capecitabine and oxaliplatin; PR, partial response; CEA, carcinoembryonic antigen.
Targeted therapy phase
During second-line chemotherapy, the patient’s CEA level decline remained suboptimal (Figure 3), indicating a limited therapeutic response (Figure 1c); consequently, the patient opted for next-generation sequencing (NGS)-based genomic profiling. On April 14, 2023, NGS gene testing was completed, revealing a DCTN1–ALK gene fusion, ATM p.E1724*, and amplification of the CDK6 gene by hybrid-capture NGS performed on DNA extracted from the gastric-biopsy formalin-fixed paraffin-embedded (FFPE) tissue on September 8, 2022. On May 3, 2023, the patient received targeted therapy with alectinib. On May 10, 2023, the patient underwent strontium-89 (Sr-89) therapy for metastatic bone tumor control and severe pain relief. During follow-up, the patient demonstrated excellent disease control: CT revealed gastric wall thinning, and the previously lytic L3 metastasis exhibited osteogenic changes (Figures 1d, e).
Post-resistance phase
In January 2024, surveillance revealed a marked surge in CEA, suggesting incipient, gradual resistance to targeted therapy. Subsequent MRI of the L3 vertebra revealed epidural spinal cord compression from osseous metastasis, prompting palliative radiotherapy to the L2–L4 metastatic lesion in February 2024. The whole blood circulating tumor DNA (ctDNA) gene test revealed the disappearance of the original DCTN1–ALK fusion and the concomitant emergence of a de novo KRAS amplification mutation. The patient subsequently experienced fulminant systemic progression and ultimately died of uncontrollable gastrointestinal hemorrhage.
Discussion
ALK rearrangements occur at low prevalence in a spectrum of non-pulmonary solid tumors, yet they have emerged as actionable, pan-cancer driver events among solid neoplasms. Activating ALK alterations—comprising gain-of-function point mutations, high-level gene amplifications, and oncogenic fusions/rearrangements—have been documented in a spectrum of malignancies that includes NSCLC, anaplastic large-cell lymphoma (ALCL; accounting for approximately 0.5% of adult lymphomas and roughly 10% of pediatric non-Hodgkin lymphomas), IMT, neuroblastoma, cutaneous spitzoid neoplasms, and inflammatory breast carcinoma. Population-level genomic profiling identifies ALK alterations in ~3.3% of all cancers, with fusions/rearrangements representing a minority subset (~0.5%–0.8%) (15). Within NSCLC, the prevalence of ALK rearrangements exceeds 3%, whereas non-NSCLC malignancies exhibit a markedly lower incidence (~0.2%). Beyond NSCLC, IMT (~50%) and ALCL (50%–80%) are the entities most frequently driven by ALK fusions. While the overwhelming majority of ALK-positive NSCLCs harbor an EML4–ALK fusion (83.5%), this chimeric transcript is encountered in only ~31% of ALK-rearranged non-NSCLC tumors, which instead display marked heterogeneity in 5′ fusion partners (16). In gastric cancer, ALK fusions reported involve RAB10 (17) or HMBOX (7); here, we report the first case of DCTN1–ALK fusion.
The appearance of the DCTN1–ALK fusion gene in cancer is a rare genetic rearrangement event, primarily associated with the occurrence and progression of tumors. DCTN1 is a protein related to the cytoskeleton, involved in the transport of materials within the cell (18). When DCTN1 undergoes a gene fusion with ALK, it may lead to the abnormal activation of the ALK kinase, thereby promoting the proliferation and survival of tumor cells. To date, the DCTN1–ALK fusion gene has been reported in three cases of NSCLC, one case of IMT (12), six cases of Spitz tumors (14), one case of epithelioid fibrous histiocytoma (19), and one case of pancreatic tumors (20).
With the advancement of NGS technology, rare ALK fusion subtypes, such as DCTN1–ALK, are being increasingly identified, which has facilitated the development of more precise, targeted treatment strategies for patients with these rare fusions. All published cases have confirmed a clinical response to targeted therapy in tumors harboring the DCTN1–ALK fusion protein. Among the three lung cases, the lung cancer patient (9) with a single DCTN1–ALK mutation received crizotinib orally at a dose of 250 mg twice daily, which resulted in a significant symptomatic improvement and radiographic response after 3 months of therapy. The female lung adenocarcinoma patient (10) (concurrent EGFR mutations and DCTN1–ALK fusion) with brain metastases developed resistance to chemotherapy or targeted therapy. CtDNA was dynamically monitored and confirmed the coexistence of a primary EGFR T790M mutation/EGFR exon 19 deletion and DCTN1–ALK translocation. She responded to osimertinib and alectinib treatment, and after acquiring osimertinib resistance, the patient still responded to alectinib and achieved a partial response (PR) for lung and brain lesions. The lung adenocarcinoma patient (11) harboring dual DCTN1–ALK and ALK-CLIP4 rearrangements showed PR to crizotinib with a 12-month progression-free survival (PFS) and received alectinib as second-line treatment. In the IMT case (12), the patient, who had confirmed a diagnosis of a DCTN1–ALK fusion through genetic profiling, was enrolled in a phase I clinical trial (ClinicalTrials.gov Identifier: NCT01548144) of crizotinib in combination with pazopanib. The patient was treated with crizotinib 250 mg orally on alternating days and pazopanib 200 mg orally daily and had confirmed PR for over 6 months. In our gastric adenocarcinoma case, the chemotherapy phase lasted for approximately 8 months. During chemotherapy, the primary gastric lesion demonstrated a limited response to chemotherapy +/− immunotherapy. However, the bone metastases exhibited primary resistance, resulting in severe pain and impairment of activities of daily living. On the contrary, alectinib targeted therapy reached PR, and the PFS reached 11.5 months.
In the other two cases of ALK fusion gastric cancer, one with RAB10–ALK mutation refused off-label use of agents such as crizotinib and ceritinib, and the other patient with ALK–HMBOX fusion received alectinib as first-line targeted therapy and achieved complete response (CR) in thoracic and cervical metastatic lymph nodes and PR in brain metastases, which reached a 6-month PFS. The second-line targeted therapy of lorlatinib also showed a 6-month PFS, with the best overall response of stable disease (SD). Likewise, other rare ALK-altered digestive system neoplasms confront therapeutic decision challenges. In a case series report of 13 GI cancer patients (7), regardless of the prior standard systemic treatment, ALKi chosen as the first-line target agent were alectinib for seven patients, crizotinib for five patients, and entrectinib for one patient. Further lines of ALKi included alectinib, lorlatinib, and ceritinib. Ten of 12 evaluable patients (83%) achieved a PR or SD response, and the median PFS was 5.0 months; the median OS was 9.3 months. Overall, patients with non-pulmonary malignancies harboring ALK alterations exhibited shorter PFS on targeted therapy than those with ALK-positive lung cancer. The breakpoint identified in our case with baseline tumor specimen (DCTN1-E19:ALK-E20) mirrors that described in NSCLC, implying that the shorter progression-free survival observed in GI malignancies does not merely depend on structural differences in the fusion itself but may be comprehensively affected by patient-related tumor microenvironment or co-occurring genomic events, such as CDK6 amplification and ATM mutation.
In summary, DCTN1–ALK fusion behaves biologically like other gastric ALK fusions [same kinase domain, equal in vitro Tyrosine kinase Inhibitor (TKI) sensitivity], but three features appear unique to the DCTN1 partner. First, DCTN1–ALK retains the same ALK kinase domain and in vitro drug sensitivity as other gastric ALK fusions, indicating equal intrinsic signaling potency. Second, it reproducibly associates with poorly differentiated histology and bone metastases, suggesting a phenotypic footprint linked to the DCTN1 cytoskeletal role. Third, our case and the published lung tumors frequently harbor co-alterations such as ATM loss or CDK6 amplification that shorten TKI durability. Taken together, the fusion is not inherently “stronger”, but its biological distinctiveness lies in histology, tropism, and genomic context, supporting comprehensive gene profiling in specific situations, especially for those with refractory gastric cancer. Therefore, based on an actionable fusion frequency ≥0.5%, a clinically relevant response rate to ALK TKIs, and marginal additional cost when incorporated into existing comprehensive NGS workflows, we advocate routine ALK fusion assessment in all cases of metastatic or refractory gastric cancer.
Dynamic genomic profiling to monitor resistance mechanisms can guide precise regimen adjustments. Given that rare variants lack evidence-based therapeutic guidance and the challenges of repeat tissue biopsies, liquid biopsy options, such as peripheral blood ctDNA testing, offer a practical alternative. Several cases have reported the use of dynamic ctDNA sequencing to identify resistance mechanisms after first-line targeted therapy, thereby informing subsequent targeted treatment selection. In the case of carcinoma of unknown primary with an EML4–ALK fusion (21), ctDNA detection revealed two resistance mutations (L1196M and G1269A) to crizotinib. Therefore, the targeted therapy was switched to brigatinib 180 mg once a day p.o., and CT examination confirmed PR. The gastric cancer case with ALK–HMBOX fusion failed to reveal the resistance mechanism by a second FoundationOne CDx test on the new cervical lymph node tissue specimen; however, the p.Val1180Leu ALKi resistance mutation was identified in ctDNA analyzed on a Illumina NextSeq 550 NGS instrument. Then, the patient started lorlatinib 50 mg twice a day as second-line treatment. After progression, an STK11 intronic loss-of-function mutation (c.734 + 1G.T) was detected as a resistance mechanism through liquid biopsy using pleural effusion and plasma. In our case, the DCTN1–ALK fusion was first detected in the primary gastric tissue specimen; cfDNA analysis was employed later for longitudinal surveillance and revealed the emergence of KRAS amplification at progression. It is supposed that the KRAS amplification mutation replaced DCTN1–ALK fusion to emerge as the driver gene, precipitating rapid and widespread tumor progression. The patient ultimately lost all further anti-tumor therapeutic opportunities. Although the index detection of DCTN1–ALK was tissue-based, subsequent resistance profiling relied on cfDNA; therefore, we cannot determine whether the KRAS-amplified sub-clone originated from the primary tumor, bone metastases, or both.
To date, a limited number of basket trials have been initiated to investigate better targeted therapy strategies and resistance mechanisms in patients with ALK-altered non-pulmonary malignancies. There are several ongoing basket trials listed on the ClinicalTrials.gov website, with identifiers NCT04644315, NCT03868423, NCT01284192, and NCT04439266 (7). With continuing advances and innovations in gene sequencing technologies, we anticipate an expansion of regional and global basket trials that will establish a robust evidence base for precision-targeted therapies against rare ALK alterations.
Conclusion
Despite the progress made in understanding and targeting ALK fusions in non-lung cancers, challenges remain in optimizing treatment strategies and overcoming resistance to ALK inhibitors. GI cancer patients harboring ALK mutations are at risk of being excluded from personalized treatment, which may dramatically improve survival due to the conventional test and treatment routine. To our knowledge, this is the first case report of gastric cancer harboring DCTN1–ALK fusion with clinical response to targeted therapy. Liquid biopsy plays a pivotal role in the dynamic genomic surveillance of patients in real-world clinical practice. Further research is needed to identify novel therapeutic combinations that can improve patient outcomes and elucidate the mechanisms of resistance to ALK-driven solid tumors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chon HJ Kim HR Shin E Kim C Heo SJ Lee C . The clinicopathologic features and prognostic impact of ALK positivity in patients with resected gastric cancer. Ann Surg Oncol. (2015) 22:3938–45. doi: 10.1245/s 10434-015-4376-8, PMID: 25707491 · doi ↗ · pubmed ↗
- 2Deng N Goh LK Wang H Das K Tao J Tan IB . A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. (2012) 61:673–84. doi: 10.1136/gutjnl-2011-301839, PMID: 22315472 PMC 3322587 · doi ↗ · pubmed ↗
- 3Shah MA Khanin R Tang L Janjigian YY Klimstra DS Gerdes H . Molecular classification of gastric cancer: A new paradigm. Clin Cancer Res. (2011) 17:2693–701. doi: 10.1158/1078-0432.CCR-10-2203, PMID: 21430069 PMC 3100216 · doi ↗ · pubmed ↗
- 4Aisner DL Nguyen TT Paskulin DD Le AT Haney J Schulte N . ROS 1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol Cancer Res. (2014) 12:111–8. doi: 10.1158/1541-7786.MCR-13-0479-T, PMID: 24296758 PMC 4140177 · doi ↗ · pubmed ↗
- 5Singhi AD Ali SM Lacy J Hendifar A Nguyen K Koo J . Identification of targetable ALK rearrangements in pancreatic ductal adenocarcinoma. J Natl Compr Canc Netw. (2017) 15:555–62. doi: 10.6004/jnccn.2017.0058, PMID: 28476735 · doi ↗ · pubmed ↗
- 6Alese OB El-Rayes BF Sica G Zhang G Alexis D La Rosa FG . Anaplastic lymphoma kinase (ALK) gene alteration in signet ring cell carcinoma of the gastrointestinal tract. Ther Adv Med Oncol. (2015) 7:56–62. doi: 10.1177/1758834014567117, PMID: 25755678 PMC 4346214 · doi ↗ · pubmed ↗
- 7Ambrosini M Del Re M Manca P Hendifar A Drilon A Harada G . ALK inhibitors in patients with ALK fusion-positive GI cancers: an international data set and a molecular case series. JCO Precis Oncol. (2022) 6:e 2200015. doi: 10.1200/PO.22.00015, PMID: 35476549 PMC 9200393 · doi ↗ · pubmed ↗
- 8Chiarle R Voena C Ambrogio C Piva R Inghirami G . The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. (2008) 8:11–23. doi: 10.1038/nrc 2291, PMID: 18097461 · doi ↗ · pubmed ↗