Prenatal Recurrence of Ductal Plate Malformations Leads to PKHD1 Variant Reclassification
Mario Abaji, Laurent Nasca, Marie‐Pierre Audrezet, Bénédicte Gerard, Xavier Vanhoye, Cécile Chau, Claude D’Ercole, Annie Levy‐Mozziconacci

TL;DR
This study shows how recurring liver abnormalities in two pregnancies helped reclassify a genetic variant as harmful, improving understanding of a liver disease gene.
Contribution
The study demonstrates how variant reclassification can occur through prenatal recurrence and multi-modal analysis.
Findings
A PKHD1 variant (c.533T>A) was reclassified as likely pathogenic due to recurrence in two pregnancies.
Combining prenatal imaging, postmortem exams, and gene sequencing improved variant classification.
The findings expand the known clinical effects of PKHD1-related disorders.
Abstract
Ductal plate malformations (DPM) encompass a spectrum of congenital liver disorders characterized by abnormal bile duct development, often associated with conditions such as Caroli disease. Variants in the PKHD1 gene cause a wide spectrum of DPM, but genotype–phenotype correlations remain challenging. We report a couple with two consecutive terminated pregnancies following prenatal detection of hepatic anomalies suggestive of DPM. Genetic analyses revealed compound heterozygous variants in PKHD1 in both fetuses. One variant (c.931A>G) was classified as likely pathogenic, while the second (c.533T>A), initially reported as a variant of uncertain significance, was reclassified as likely pathogenic after recurrence of the phenotype. This case highlights the importance of integrating prenatal imaging, postmortem examination, and whole‐gene sequencing to refine variant classification and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
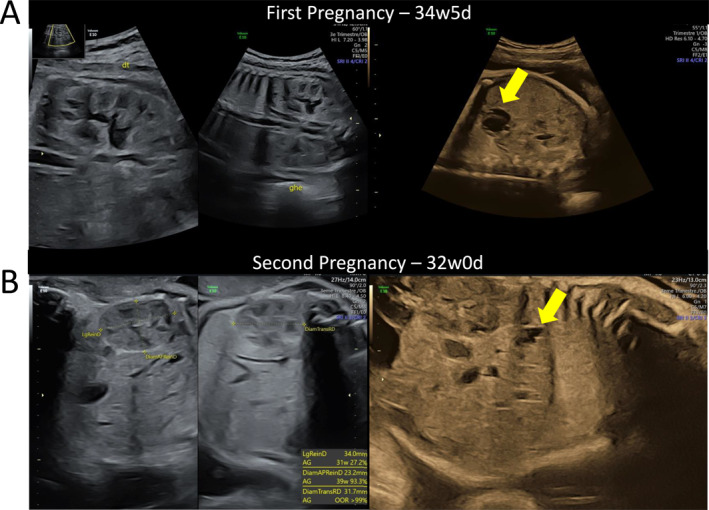 FIGURE 1
FIGURE 1| Case | Parental details | Gestation at diagnosis | Phenotypes (HPO terms) | Obstetric history | Family history | Outcome | ||
|---|---|---|---|---|---|---|---|---|
| First pregnancy | Maternal | Age | 34 yo | 34 GA and 5 days |
Malformation of the hepatic ductal plate (HP:0006563) Renal cortical microcysts (HP:0004734) Ventricular septal defect (HP:0001629) Left ventricular hypertrophy (HP:0001712) Dysplastic tricuspid valve (HP:0030732) Short philtrum (HP:0000322) | G1P0 | No significant family history | Termination of pregnancy at 38 GA and 3 days |
| Ethnicity | European | |||||||
| Paternal | Age | 31 yo | ||||||
| Ethnicity | European | |||||||
| Second pregnancy | Maternal | Age | 38 yo | 33 GA and 1 day | Malformation of the hepatic ductal plate (HP:0006563) | G2P0 | Termination of pregnancy at 35 GA and 2 days | |
| Paternal | Age | 34 yo | ||||||
| Variant | Zygosity | Initial criteria applied | Initial ACMG classify‐cation | Added criteria following the recurrence | Final classification | |
|---|---|---|---|---|---|---|
| Paternal allele | Heterozygote | PM2_moderate, PP3_supporting, PP4_moderate, PP5_supporting, PM5_moderate | Likely pathogenic (class IV) | PP1_ supporting | Likely pathogenic (class IV) | |
| Maternal allelle | Heterozygote | PM2_moderate, PM3_moderate, PP4_supporting | Uncertain significance (class III) |
PP1_supporting PP2_Absence of other variation detected by genome sequencing | Likely pathogenic (class IV) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Kidney Cyst Diseases · Renal and related cancers · Genetic Syndromes and Imprinting
Fetal Phenotype
1
A couple was referred to the prenatal diagnosis center, following the detection of ultrasound anomalies in two consecutive pregnancies after an uneventful first two trimesters (Table 1A).
First Pregnancy
1.1
Routine third‐trimester ultrasound at 33 years of gestational age (GA) revealed an enlarged right adrenal gland, confirmed at the prenatal center (Figure 1A). Fetal MRI clarified the hepatic origin, identifying multiple cystic lesions, some with a tubular configuration, affecting both liver lobes. Due to the poor prognosis, the couple opted for termination of pregnancy (TOP). Postmortem examination confirmed a female fetus with a large multi‐cystic hepatic lesion, ductal plate malformation (DPM), right renal hypertrophy with possible tubular cysts, congenital heart disease and dysmorphic features.
Prenatal ultrasound images from the two consecutive pregnancies showing bilateral kidneys on the left side and the liver on the right side. (A) First pregnancy at 34 + 5 GA: heterogeneous, hepatic non‐vascularized cystic‐solid mass (32 × 23 mm, arrow) (B) second pregnancy at 32 GA: multiple hepatic anechoic lesions (largest 12 × 4 mm, arrow). Kidneys increase in volume.
Second Pregnancy
1.2
At 32 GA, ultrasound detected multiple hepatic anechoic lesions (Figure 1B), confirmed on MRI at 33 GA with a diffuse arborizing pattern and mild renal enlargement. TOP was performed and postmortem examination confirmed a female fetus with isolated DPM.
Diagnostic Method
2
First Pregnancy
2.1
Amniocentesis was performed before TOP at 38 weeks of gestation. Chromosomal microarray analysis (CMA) was normal, and targeted sequencing for hepatorenal polycystic disease genes was conducted (Supporting Information S1).
Second Pregnancy
2.2
Amniocentesis at 33 GA revealed a normal CMA. The same targeted analysis was performed followed by quad exome sequencing, in addition to whole gene analysis of PKHD1 with short read sequencing (Supporting Information S1).
Diagnostic Results and Interpretation
3
In the first pregnancy, genetic testing using the targeted gene panel for hepatorenal polycystic disease identified two PKHD1 variants inherited in trans. The first, c.931A>G (p.(Thr311Ala)) [1], was classified as likely pathogenic. The second, c.533T>A (p.(Val178Glu)), was initially designated as a variant of uncertain significance (VUS). After recurrence of the hepatic phenotype in the second pregnancy and identification of the same two variants, along with the exclusion of other causative variants through exome and PKHD1 whole gene sequencing, the c.533T>A variant was reclassified as likely pathogenic (Table 1B).
Discussion
4
Ductal plate malformations (DPM) encompass a spectrum of congenital liver disorders including Caroli disease, a non‐obstructive dilatation of the intrahepatic bile ducts. Variants in PKHD1, primarily associated with autosomal recessive polycystic kidney disease (ARPKD), can also lead to a broad range of phenotypes, from asymptomatic cases to severe liver and/or kidney involvement with perinatal lethality. This phenotypic variability complicates genotype‐phenotype correlations and prenatal diagnosis [2, 3, 4, 5].
In this report, two consecutive pregnancies were terminated due to DPM. Genetic analysis revealed compound heterozygous PKHD1 variants in both fetuses. Initially, one variant was classified as likely pathogenic and the other as a variant of uncertain significance (VUS). Reclassification of the second variant was supported by: (i) recurrence of the phenotype within the family; (ii) the high clinical specificity associated with PKHD1 and the absence of known genetic heterogeneity for this condition; and (iii) genomic short‐read sequencing of the gene, which did not reveal any additional variants. Although the absence of additional findings does not definitively exclude complex variations that may not be detectable by short‐read sequencing (e.g., repeat expansions, large insertions/deletions, or variants in low‐mappability regions), it significantly lowers the likelihood of another pathogenic variant. This absence of additional variation may be considered an additional supporting criterion in the ACMG framework when interpreting recessive variants in genes with strong phenotype specificity.
Although congenital heart malformations have not been previously described in association with PKHD1 mutations, children with ARPKD frequently exhibit cardiac abnormalities, including left ventricular geometric alterations and systolic dysfunction [6]. This case expands the spectrum of PKHD1‐related disorders. Thanks to this work, non‐invasive prenatal testing for the paternal PKHD1 variant was performed in the subsequent pregnancy (Supporting Information S2: pedigree chart), confirming that the fetus was not a carrier and resulting in the birth of a healthy child.
Ethics Statement
This study was approved by the Ethics and Scientific Committee (Comité d’Éthique et Scientifique, CSE) of AP‐HM under approval number CSE25‐95. The committee reviewed the project on March 12, 2025, and issued a favorable opinion after evaluating its scientific rationale, feasibility, patient information process, and ethical considerations.
Consent
The couple consented for publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting Information S1
Supporting Information S1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1P. J. Freeman , R. K. Hart , L. J. Gretton , A. J. Brookes , and R. Dalgleish , “Variant Validator: Accurate Validation, Mapping, and Formatting of Sequence Variation Descriptions,” Human Mutation 39, no. 1 (2018): 61–68, 10.1002/humu.23348.28967166 PMC 5765404 · doi ↗ · pubmed ↗
- 2C. Giacobbe , F. Di Dato , D. Palma , M. Amitrano , R. Iorio , and G. Fortunato , “Rare Variants in PKHD 1 Associated With Caroli Syndrome: Two Case Reports,” Molecular Genetics & Genomic Medicine 10, no. 8 (2022): e 1998, 10.1002/mgg 3.1998.35715958 PMC 9356553 · doi ↗ · pubmed ↗
- 3J. Courcet , A. Minello , F. Prieur , et al., “Compound Heterozygous PKHD 1 Variants Cause a Wide Spectrum of Ductal Plate Malformations,” American Journal of Medical Genetics ‐ Part A 167, no. 12 (2015): 3046–3053, 10.1002/ajmg.a.37352.26385851 · doi ↗ · pubmed ↗
- 4Wei Y. , Cao J. , Xing C. , et al., “Prenatal Diagnosis of Caroli’s Disease by Ultrasound and MRI Imaging,” Prenatal Diagnosis 17, no. 4 (2025): 6750, 10.1002/pd.6750.39821604 · doi ↗ · pubmed ↗
- 5C. Bergmann , J. Senderek , F. Schneider , et al., “ PKHD 1 Mutations in Families Requesting Prenatal Diagnosis for Autosomal Recessive Polycystic Kidney Disease (ARPKD): PKHD 1 In Families Requesting PD for ARPKD,” Human Mutation 23, no. 5 (2004): 487–495, 10.1002/humu.20019.15108281 · doi ↗ · pubmed ↗
- 6M. Chinali , L. Lucchetti , A. Ricotta , et al., “Cardiac Abnormalities in Children With Autosomal Recessive Polycystic Kidney Disease,” Cardiorenal Medicine 9, no. 3 (2019): 180–189, 10.1159/000496473.30844805 · doi ↗ · pubmed ↗