Spontaneous Pregnancy in Genetically Confirmed 11-Beta Hydroxylase Deficiency: A Case Series and Literature Review
Pushpa Machineni, Asha Ranjan, Adlyne Reena Asirvatham, Kunal Gupta, Shriraam Mahadevan

TL;DR
Two women with genetically confirmed 11-beta hydroxylase deficiency achieved spontaneous pregnancies, highlighting the condition's varied effects on fertility.
Contribution
Reports two rare cases of spontaneous pregnancy in genetically confirmed 11βOHD, expanding understanding of its reproductive outcomes.
Findings
A woman with classic 11βOHD had a successful spontaneous pregnancy despite high androgen levels and prior surgery.
A non-classic 11βOHD patient experienced multiple spontaneous pregnancies and showed a broad range of clinical features.
The cases emphasize the importance of accurate diagnosis and reproductive counseling for CAH patients.
Abstract
11-beta hydroxylase deficiency (11βOHD) is a rare variant of congenital adrenal hyperplasia (CAH) with autosomal recessive inheritance, resulting in androgen excess and mineralocorticoid precursor accumulation. Fertility is often impaired due to hyperandrogenism and anatomical abnormalities, and spontaneous successful pregnancy in classic form is rare. We describe two cases. Case 1 is a 27-year-old woman with genetically confirmed classic 11βOHD (homozygous CYP11B1 splice-site variant c.240-2A>G), initially misdiagnosed as 21-hydroxylase deficiency, who achieved spontaneous conception despite high androgen levels, long-term steroid exposure, and prior genital surgery and delivered a healthy male child. Case 2 is a genetically confirmed non-classic 11βOHD (homozygous, missense variation, c.412C>T), presented with medullary nephrocalcinosis, clinical history of polycystic ovary syndrome…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
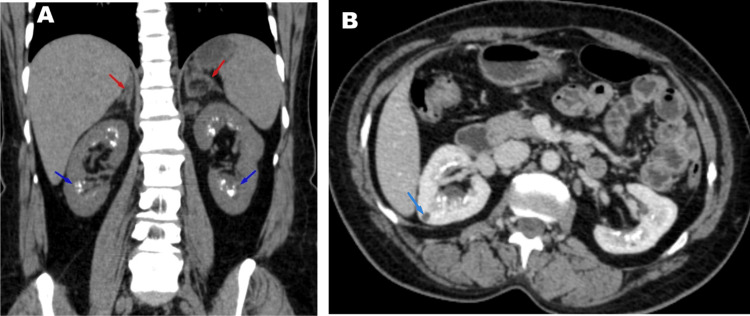 Figure 1
Figure 1| Hormone/Test | Case 1 Result (Reference range) | Case 2 Result (Reference range) |
| Adrenocorticotropic hormone (ACTH) (pmol/L) | — | 74 (<10.1) |
| Cortisol (nmol/L) | 109(132–537) | 219 (132–537) |
| 17-Hydroxyprogesterone (nmol/L) | 28.7(0.6-3.9) | 249.3 (ACTH Stimulated) (6–66.5)a |
| 11-Deoxycortisol (nmol/L) | 41.5(<3.1) | 546.1 (1.5-8.7)a |
| 11-Deoxycorticosterone (nmol/L) | — | 64.1 (0.06-0.44)a |
| Androstenedione (nmol/L) | — | 63 (1.04-12.2)a |
| Total Testosterone(nmol/L) | 12.1(0.7-2.8) | 8.4(0.7-2.8)a |
| Dehydroepiandrosterone sulfate(DHEAS) (µmol/L) | 0.49(1.6-9.1) | 15.98(1.6-9.1) |
| Sodium (mEq/L) | 138(135–145) | 141 (135–145) |
| potassium (mEq/L) | 3 (3.5–5.0) | 2.9 (3.5–5.0) |
| Bicarbonate(mEq/L) | 21(21-31) | 22(21-31) |
| Chloride(mEq/L) | 101(101-109) | 103(101-109) |
| Calcium (mmol/L) | — | 2.35(2.15-2.50) |
| Phosphorus (mmol/L) | — | 1.29 (0.81-1.45) |
| Vitamin D (nmol/L) | — | 55 (>50) |
| Parathyroid hormone (pmol/L) | — | 3.6(1.6-6.9) |
| Creatinine(µmol/L) | 52(44 – 80) | 60(44 – 80) |
| Parameter | Classic 11β-OHD | Non-classic 11β-OHD | |||
| Case 1 | Simm et al., 2007 [ | Kavitha et al., 2023 [ | Demir et al., 2021 [ | Case 2 | |
| Genetic Mutation | Homozygous c.240-2A>G | Heterozygous intron DS+2 splicing + exon 8(G444D) | NR | Homozygous p.L299P (c.896T>C) | Homozygous c.412C>T |
| Clinical Presentation | Ambiguous genitalia at birth | Ambiguous genitalia at birth | Ambiguous genitalia | Ambiguous genitalia at birth | PCOS-like, short stature, Hypertension with hypokalemia |
| Surgical History | Clitoroplasty + vaginoplasty | Clitoral reduction + introitoplasties. | Clitoral resection + vaginoplasty + dilation | Surgeries at 4 and 24 years | None |
| Hypertension | Yes | No | Yes | Yes | Yes |
| Mode of Conception | Spontaneous | Clomiphene-induced | NR | Spontaneous | Spontaneous |
| Steroid Regimen | Prednisolone 7.5mg | Dexamethasone ↑ to 2 mg/day | Prednisolone | Methylprednisolone + Dexamethasone → Hydrocortisone | None (prenatal/natal) |
| Pregnancy Outcome | Uneventful. Elective C-section at 38weeks.Male child. | GHTN. Hyperandrogenism. Elective C-section at 37 weeks. Male child. | Severe preeclampsia. Emergency C-section at 33 weeks. Male child. | Elective C-section at 34 weeks. Female child. | Uneventful. Elective C-section at 37 weeks, Male and female child |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSexual Differentiation and Disorders · Genetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Hormonal and reproductive studies
Introduction
11-beta hydroxylase deficiency (11βOHD) is a rare variant of congenital adrenal hyperplasia (CAH), resulting from mutations in the CYP11B1 gene. The enzymatic defect leads to the accumulation of 11-deoxycortisol and deoxycorticosterone, and excess production of adrenal androgens. Clinically, it is characterized by hypertension, virilization, and glucocorticoid deficiency. The classic form is typically present in infancy with virilization in female infants, and the non-classic form manifests later in childhood or adolescence with signs of androgen excess such as hirsutism and oligomenorrhea in females [1]. Impaired fertility may be due to both hormonal and anatomical factors. Elevated adrenal androgens impair folliculogenesis, ovulation, and endometrial receptivity. Concurrently, increased adrenal progesterone secretion alters gonadotropin-releasing hormone (GnRH) and luteinizing hormone (LH) pulsatility, impairs cervical mucus quality, and disrupts endometrial development and implantation. Structural abnormalities from prior genital reconstructive surgeries may further compromise reproductive potential [2].
Case presentation
Case 1 is a 27-year-old female, born of a second-degree consanguineous marriage, who had ambiguous genitalia at birth with no hyperpigmentation and adrenal crisis. Clinical examination revealed clitoromegaly, fused labia minora with a single urogenital opening-Prader stage III. Karyotype was 46, XX. Biochemical evaluation revealed elevated 17-hydroxyprogesterone (Table 1). Pelvic ultrasound showed a normal uterus with no adrenal hyperplasia. A presumptive diagnosis of 21-hydroxylase deficiency was made, and hydrocortisone was initiated. Fludrocortisone was added subsequently. At six months of age, the patient underwent recession clitoroplasty and vaginoplasty. She attained menarche at age 15 with normal secondary sexual characteristics and had regular menstrual cycles. However, she had persistent hypokalemia with no history of hypertension, and at the age of 17, after discontinuing glucocorticoid therapy for re-evaluation, she developed an episode of hypokalemic paralysis. Biochemical investigation at that time revealed elevated 11-deoxycortisol (Table 1), and dexamethasone was restarted. She was later diagnosed with hypertension, which was well controlled with glucocorticoid therapy. During the course, her androgen levels were well controlled. Genetic testing confirmed CYP11B1 homozygous splice-site variant mutation (c.240-2A>G), establishing the diagnosis of classic 11β-OHD. She conceived spontaneously; during pregnancy, she was shifted to prednisolone 7.5mg per day. She had an uneventful antenatal course (no gestational diabetes mellitus (GDM)) and delivered a healthy male child.
Case 2 is a 37-year-old female, born of a second-degree consanguineous marriage, who had normal female genitalia at birth, with a positive family history of 11β-OHD in her nephews. At age 10, she developed hyperpigmentation, hirsutism, and menstrual irregularities but was not evaluated. Later, she developed hypertension and hypokalemia, which remained unevaluated for many years. She had two spontaneous, uncomplicated pregnancies, delivered one female and one male child. A Computed Tomography (CT) scan done later for unrelated reasons revealed bilateral adrenal hyperplasia and medullary nephrocalcinosis with renal cysts (Figure 1A, 1B). On examination, she had short stature (height: 140 cm, -3.2 SDS), hirsutism, clitoromegaly, and hyperpigmentation. With the above findings, CAH was suspected and evaluated further. Biochemical and steroid profiling are presented in Table 1. Steroid profiling revealed elevated 17-hydroxyprogesterone and 11-deoxycortisol (546.1 (1.5-8.7)), consistent with the diagnosis of CAH. Evaluation for medullary nephrocalcinosis revealed normal serum calcium, phosphorus, parathyroid hormone, and urinary calcium levels, with no evidence of metabolic acidosis. Genetic testing revealed a homozygous missense variant mutation in CYP11B1: c.412C>T (p. Arg138Cys), confirming the diagnosis of non-classic 11β-OHD. She was initiated on dexamethasone, following which her androgen levels showed a progressive decline, and menstrual cycles normalized over subsequent months. Blood pressure remained within the normal range on follow-up without the need for additional antihypertensive agents.
CT scan findings of Case 2: non-classic 11-beta hydroxylase deficiencyA: Bilateral adrenal hyperplasia with myelolipoma (red arrow). Medullary nephrocalcinosis (blue arrow)B: Renal cyst with medullary nephrocalcinosis
Discussion
CAH is most commonly caused by 21-hydroxylase deficiency, while 11β-OHD accounts for approximately 5-8% of cases and has an estimated incidence of one in 100,000 live births [3]. This condition results from mutations in the CYP11B1 gene, leading to defective conversion of 11-deoxycortisol and deoxycorticosterone to cortisol and corticosterone, respectively. The biochemical consequences include cortisol deficiency, accumulation of 11-deoxycorticosterone (a potent mineralocorticoid), elevated 11-deoxycortisol, and excess adrenal androgens. Clinically, this manifests as hypertension (with suppressed renin), hypokalemia, hyperandrogenism, and genital ambiguity. Females with the classic form typically present at birth with ambiguous external genitalia and normal internal reproductive anatomy, often requiring early surgical correction. Phenotypic variability depends on residual enzymatic activity [4]. Our patient with the classic form of 11β-OHD presented in infancy with ambiguous genitalia consistent with in utero androgen excess. In our case, initial elevation of 17-hydroxyprogesterone led to a presumptive diagnosis of 21-hydroxylase deficiency, a common diagnostic pitfall due to overlapping features with 11β-OHD. Upon withdrawal of glucocorticoids, the patient experienced an episode of hypokalemic paralysis, which was likely secondary to unopposed deoxycorticosterone-mediated mineralocorticoid activity. Though elevated 17-hydroxyprogesterone levels most commonly suggest 21-hydroxylase deficiency, it is crucial to consider less common conditions such as 11β-OHD in the differential diagnosis of CAH. Similar clinical courses have been reported in the literature [5]. Hypertension and hypokalemia are characteristic features of 11β-OHD, distinguishing it from 21-hydroxylase deficiency, which typically presents with hypotension and hyperkalemia. However, the markedly elevated 11-deoxycortisol level and the identification of a homozygous CYP11B1 splice-site mutation (c.240-2A>G) ultimately confirmed 11β-OHD in this case. This diagnostic delay highlights the importance of specific steroid precursor profiling in all cases of CAH, particularly when hypertension or hypokalemia is present. Our patient had normal secondary sexual characteristics and regular menstrual cycles while on appropriate glucocorticoid replacement, suggesting adequate adrenal androgen control. Despite the well-known challenges to fertility in 11β-OHD, owing to elevated follicular-phase progesterone, gonadotropin suppression, and anatomical alterations, she achieved spontaneous conception. She was well maintained on prednisolone throughout pregnancy without any hypertensive events. This case emphasizes the potential for fertility in classic 11β-OHD when managed appropriately. Only a few genetically confirmed severe classic forms of 11β-OHD cases with successful pregnancy have been reported. A comparison with the literature (Table 2) reveals further heterogeneity. Simm et al. [6] reported a patient requiring ovulation induction with clomiphene, hyperandrogenism, and intensified glucocorticoid therapy during pregnancy. Kavitha et al. [7] described a patient on prednisolone and antihypertensives whose pregnancy was complicated by severe preeclampsia, necessitating emergency cesarean at 33 weeks. Demir et al. [8] also reported spontaneous conception following multiple reconstructive surgeries, with glucocorticoid adjustment during pregnancy. In pregnant women with 11β-OHD, hypertension is managed with glucocorticoids (hydrocortisone or prednisolone) to suppress adrenocorticotropic hormone (ACTH) driven mineralocorticoid excess. If additional therapy is needed, pregnancy-safe antihypertensives such as labetalol or nifedipine may be added, while angiotensin-converting enzyme inhibitors, angiotensin receptor II blockers, and thiazides are avoided [3,9]. During pregnancy, dexamethasone should be avoided because placental 11β-hydroxysteroid dehydrogenase type 2 does not inactivate it, allowing significant transplacental passage; and stress dose glucocorticoids are recommended during labour [10]. Cesarean section was required in 52-84% of cases, primarily due to a history of genital reconstructive surgery. Although GDM is a recognized complication in CAH pregnancies, our patient did not develop GDM. Given the limited literature on pregnancies in women with 11β-hydroxylase deficiency, clinical management is mostly guided by experience from the more prevalent 21-hydroxylase deficiency [4,11].
In contrast, the non-classic 11β-OHD patient had a much subtler phenotype and was diagnosed in adulthood despite early signs of hyperandrogenism, short stature, and menstrual irregularities from childhood. The normal appearance of external genitalia at birth and lack of salt-wasting symptoms delayed recognition, as is frequently seen in non-classic 11β-OHD. These patients are often misdiagnosed with polycystic ovary syndrome (PCOS) [4,12,13], and diagnosis requires a high index of suspicion, especially when accompanied by persistent hypertension and unexplained hypokalemia. In this case, significantly elevated levels of 11-deoxycortisol and deoxycorticosterone, along with ACTH elevation and genetic confirmation of a homozygous missense mutation in CYP11B1 (c.412C>T; p. Arg138Cys), were diagnostic. Imaging revealed bilateral adrenal hyperplasia, consistent with the underlying enzymatic defect. Additionally, she was found to have renal cysts and medullary nephrocalcinosis, most likely a consequence of long-standing chronic hypokalemia. Similar renal manifestations in patients with 11β-OHD have been previously reported by Aswani et al. and Abdulla et al. [14,15]. Remarkably, she achieved spontaneous pregnancies despite the absence of treatment, highlighting the broad clinical variability of non-classic 11β-OHD. Timely recognition and genetic confirmation are critical to prevent long-term complications and optimize reproductive outcomes.
Conclusions
These cases illustrate the broad clinical spectrum of 11β-OHD and the importance of distinguishing it from 21-hydroxylase deficiency through 11-deoxycortisol (and deoxycorticosterone when possible) measurement and genetic confirmation. Hypertension and hypokalemia are key diagnostic clues, and long-term complications like nephrocalcinosis require surveillance. Fertility is achievable with appropriate glucocorticoid therapy and management of mineralocorticoid and androgen excess. Increased awareness of the non-classic presentation is crucial to prevent diagnostic delays and associated morbidity.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rare types of congenital adrenal hyperplasias other than 21-hydroxylase deficiency J Clin Res Pediatr Endocrinol İsakoca M ErdeveŞ Çetinkaya S 23321720253971388410.4274/jcrpe.galenos.2024.2024-6-21-SPMC 11730102 · doi ↗ · pubmed ↗
- 2Pregnancy in congenital adrenal hyperplasia Endocrinol Metab Clin North Am Reisch N Auchus RJ 3914075320243908481510.1016/j.ecl.2024.05.005 · doi ↗ · pubmed ↗
- 3Steroid 11 beta-hydroxylase deficiency and related disorders Endocrinol Metab Clin North Am White PC 613020011134493910.1016/s 0889-8529(08)70019-7 · doi ↗ · pubmed ↗
- 4Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency Endocrine Bulsari K Falhammar H 19365520172792872810.1007/s 12020-016-1189-x · doi ↗ · pubmed ↗
- 5Congenital adrenal hyperplasia with 11 beta-hydroxylase deficiency J Formos Med Assoc Chang SH Lee HH Wang PJ Chen JH Chu SY 8608641032004 https://pubmed.ncbi.nlm.nih.gov/15549155/15549155 · pubmed ↗
- 6Successful pregnancy in a patient with severe 11-beta-hydroxylase deficiency and novel mutations in CYP 11B 1 gene Horm Res Simm PJ Zacharin MR 2942976820071772633310.1159/000107651 · doi ↗ · pubmed ↗
- 7Pregnancy in a woman with congenital adrenal hyperplasia with 11-beta-hydroxylase deficiency: a case report Obstet Med Krishnan K Pillai S Vaidyanathan G 66681620233713950410.1177/1753495 X 211042729 PMC 10150300 · doi ↗ · pubmed ↗
- 8Successful spontaneous pregnancy and successful delivery in a patient with severe 11-beta hydroxylase deficiency EA Suat DA Tufekci D Emur GY 222642021