Mutational Landscape and Clinical Impact of SPEN Mutations in Patients with Chronic Lymphocytic Leukemia
Priyatharsini Nirmalanantham, Andrés E. Quesada, Anindita Ghosh, Pei Lin, Chi Y. Ok, Richard K. Yang, Hong Fang, Sofia Garces, Rashmi Kanagal-Shamanna, Sanam Loghavi, Mark J. Routbort, Cameron Cheng Yin, Wang Wei, Sarah Pasyar, Roland Bassett, Siba El Hussein, Nitin Jain

TL;DR
This study finds that SPEN mutations in chronic lymphocytic leukemia are linked to worse patient outcomes and should be considered in diagnostic testing.
Contribution
The study identifies SPEN mutations as a prognostic marker in CLL and reports their frequency and associated clinical features in a large patient cohort.
Findings
SPEN mutations are found in 2.9% of CLL patients and are associated with shorter time-to-first treatment.
SPEN-mutated patients show higher frequencies of IGHV unmutated status, CD38 positivity, ZAP70 positivity, and trisomy 12.
NOTCH1 mutations are the most common co-occurring alterations with SPEN mutations in CLL.
Abstract
SPEN mutations have been described in a limited number of CLL cases and previous studies have suggested that mutations in the NOTCH1 regulatory pathway including SPEN are associated with adverse patient outcomes. However, the clinicopathologic features including the molecular landscape of SPEN mutations in CLL patients have not been extensively studied. We conducted this study to assess the frequency of SPEN mutations and their potential clinical impact on outcomes in a large cohort of CLL patients. We also describe co-occurring gene alterations in this cohort. We show in this study that SPEN mutations are associated with shorter time-to-first treatment in CLL patients. Background/Objectives: NOTCH1 is frequently mutated in chronic lymphocytic leukemia (CLL) and is a marker of poor prognosis. In addition to NOTCH1, mutations in the NOTCH1 regulatory pathway including SPEN have been…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
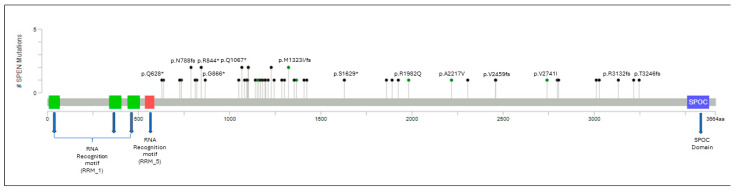 Figure 1
Figure 1 Figure 2
Figure 2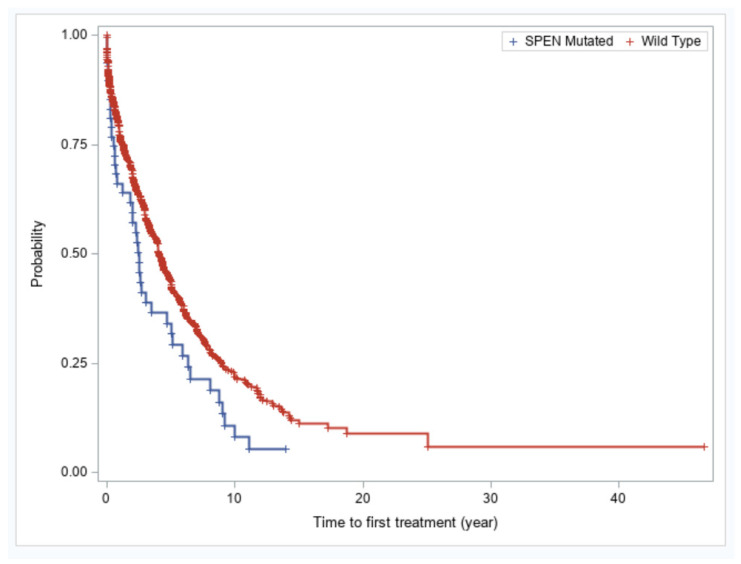 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChronic Lymphocytic Leukemia Research · Renal Diseases and Glomerulopathies · Chronic Myeloid Leukemia Treatments
1. Introduction
Chronic lymphocytic leukemia (CLL) is the most frequent leukemia affecting adults in the western world [1,2]. Despite increased understanding of CLL pathogenesis and the introduction of new therapies, CLL remains an incurable disease. CLL patients can have diverse clinical courses: some patients have an indolent course whereas others require therapy shortly after diagnosis. Many clinicopathologic features have been shown to have value in predicting prognosis, including clinical stage, expression of CD38 and/or ZAP70, somatic mutations of the immunoglobulin heavy chain variable (IGHV) genes, identification of chromosomal abnormalities by conventional cytogenetics or fluorescence in situ hybridization, including deletions of chromosome loci 13q14, 17p13, and 11q23, and trisomy 12 [3,4,5,6]. More recently, in depth genomic characterization of CLL cases has shown great heterogeneity at the molecular level. A number of genes are recurrently mutated in CLL, and, in particular, TP53, NOTCH1, and SF3B1 are associated with adverse clinical outcomes [7,8,9,10,11].
NOTCH1 is a cell surface receptor that releases its intracellular domain as a transcription factor upon activation, and plays key roles in cell proliferation, differentiation, and apoptosis. After binding to its ligand, NOTCH1 undergoes two successive proteolytic cleavages resulting in the release of its active form, which translocates into the nucleus, thereby mediating transcriptional activation of target genes including TP53, MYC, and components of the nuclear factor kB (NF-kB) pathway. NOTCH1 signaling has been shown to be important in CLL pathogenesis and is one of the most frequently mutated genes associated with advanced stage at diagnosis and poorer overall survival [9].
Several proteins negatively regulate NOTCH1 activity, including the tumor repressors F-box/WD repeat-containing protein 7 (FBXW7), mediator complex subunit 12 (MED12), and SPEN family transcriptional repressor (SPEN) [9,11,12]. Whereas FBXW7 and MED12 act to prevent proteasomal degradation of NOTCH1, SPEN directly interferes with NOTCH1 signaling. SPEN encodes for an adaptor protein that is a part of the Mint/SHARP/SPEN complex. This complex interacts with several transcription factors, including Msx2 and RBP-J, and acts as a bridge between RBP-J and repressor proteins, such as NCor/SMRT/HDACs. RBPJ is essential for DNA binding of the NOTCH1 intracellular domain (NICD1). SPEN functions as a co-repressor of RBPJ, the nuclear effector of the Notch pathway [9,11,12]. Mutations impacting the function of FBXW7, MED12, and SPEN, by inactivating their repressor function, result in enhanced NOTCH1 signaling which may influence patient outcomes via NOTCH1 activation.
Inactivating SPEN mutations have been reported in a limited number of CLL cases assessed with a frequency ranging from approximately 1% to 9% [12,13,14,15]. Others have reported that mutations in the NOTCH1 regulatory pathway including SPEN predict a shorter time-to-first treatment in a small cohort of CLL patients [15]. We conducted this study to assess the frequency of SPEN mutations and their potential clinical impact on outcomes in a large cohort of CLL patients. We also describe co-occurring gene alterations in this cohort.
2. Materials and Methods
2.1. Patients and Samples
Samples of peripheral blood and bone marrow aspirates from 1617 CLL patients were subjected to targeted sequencing using a 29-gene panel (EndCLL Assay V1). The percentage of CLL cells in each sample was estimated by multi-color flow cytometry immunophenotypic analysis. Only samples that contained at least 10% monotypic CD5+/CD19+ B cells underwent sequencing with the EndCLL Assay V1.
2.2. Clinical and Laboratory Characteristics
Demographic, clinical, and laboratory data included age, gender, prior treatments, clinical follow-up, flow cytometry immunophenotypic findings, expression of ZAP70 and CD38, IGHV mutation status, and fluorescence in situ hybridization (FISH) results. These data were evaluated and collected at the time (±60 days) of next generation sequencing sample collection. Static prognostic factors, such as IGHV mutation status and ZAP70 expression, were not repeated at time of sample collection for NGS if already known. These data were extracted from the medical records using an Institutional Review Board-approved chart review protocol.
2.3. Targeted Next Generation Gene Sequencing
The gene panel employed included 29 genes known to be mutated in CLL as has been described in [16]. This panel included the following: ATM, BIRC3, BTK, CALR, CARD11, CD79A, CD79B, CHD2, CSMD3, CXCR4, DDX3X, EZH2, FAT1, FBXW7, KLHL6, LRP1B, MAPK1, MUC2, MYD88, NOTCH1, PLCG2, PLEKHG5, POT1, SF3B1, SPEN, TGM7, TP53, XPO1, and ZMYM3. Briefly, sequencing libraries for target regions in 29 genes were prepared from 250 ng of genomic DNA isolated from peripheral blood or bone marrow samples. The Agilent Haloplex HS target enrichment system (Agilent Technologies, Santa Clara, CA, USA) was utilized for the library preparation. Incorporation of molecular barcodes in the DNA library allowed removal of duplicate reads and improved sensitivity of detection at low variant allelic frequencies (VAF), a useful design feature suitable for the detection of subclonal mutations in CLL. Paired-end bidirectional sequencing was performed using the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA). A minimum average depth of 3000× per sample, and a minimum of 80% reads at a quality score of AQ30 or better were required for the interpretation. Paired germline testing was not performed due to challenges of obtaining a reliable germline sample (i.e., cultured skin fibroblasts) for routine testing in hematological malignancies. Variant calling was performed using Agilent SureCall software 4.2.2 (Agilent Technologies) and for variants not listed in single nucleotide polymorphism databases, we took into account the tumor cell percentage, cytogenetics data for chromosomal changes, VAF of novel variant, and VAF of known somatic mutation to identify variants of potential germline variants. An internal database of all the variants along with the distribution of VAF to identify potential germline variants was employed. Variants with heterozygous or homozygous VAF without supporting literature confirming somatic origin or without the tumor percentage and VAF data supporting somatic origin were reported separately from the known somatic mutations as variants of uncertain origin and removed from the final analysis/cohort. Final review and reporting of the results were performed by a pathologist.
2.4. Statistical Analyses
The patient baseline characteristics were summarized using descriptive statistics. Wilcoxon rank-sum and Chi-square tests were used to assess the association between SPEN mutational status and continuous and categorical variables, respectively. The Kaplan–Meier method was used to estimate the time to starting first treatment from the diagnosis date and the log-rank test was used to compare the different groups. All statistical analyses were performed using SAS version 9.4. All statistical tests used a significance level of 5%. The association between time-to-first treatment and known risk factors were evaluated using univariate and multivariate Cox proportional hazard models.
3. Results
3.1. Patients and Clinical Characteristics
The CLL patients included in this study are summarized in Table 1, according to SPEN mutational status. Overall, we detected SPEN mutations in 48 of 1617 (2.9%) CLL cases. The median age of patients at diagnosis of SPEN mutated CLL was 65 years (range, 40–87) with a male to female ratio of 1.3:1. There were 31 (64.6%) treatment-naïve and 17 (35.4%) previously treated patients (relapsed/refractory). At the time of data collection, 8 (16.7%) patients had died. The median follow-up interval was 9.9 years.
3.2. SPEN Mutations
A total of 53 individual SPEN mutations were identified in 48 CLL cases. A detailed summary of these mutations is shown in Table 2, Figure 1 and Supplementary Table S1. In 10 (21%) patients, >1 SPEN mutation was detected. A total of 49 of 53 (92.4%) SPEN mutations were deleterious (frameshift or truncating nonsense mutations) with the remaining 4 (7.6%) mutations being missense. SPEN mutations were located toward the carboxy-terminal, upstream of the SPEN paralog and ortholog C-terminal (SPOC) domain.
3.3. Concurrent Gene Mutations
Concurrent gene mutations detected in the 48 CLL cases with SPEN mutations included the following: 21 (43.7%) NOTCH1, 11 (22.9%) TP53, 6 (12.5%) BIRC3, 5 (10.4%) SF3B1, 4 (8.3%) XPO1, 3 (6.2%) MUC2, 2 (4.2%) ATM, 2 (4.2%) FBXW7, 2 (4.2%) BTK, 1 (2.1%) POT1, 1 (2.1%) ZMYM3, 1 (2.1%) MYD88, 1 (2.1%) CXCR4, and 1 (2.1%) LRP1B (Figure 1). FAT1, DDX3X, CALR, PLCG2, and CARD11 were not mutated in cases of CLL with SPEN mutation (Figure 2).
3.4. Correlation with Other CLL Prognostic Biomarkers
Compared with CLL patients with SPEN wild type, CLL patients with SPEN mutations had a statistically higher frequency of IGHV unmutated status (79.5% vs. 57.8%, p = 0.004), CD38 positivity (73.3% vs. 52.4%, p = 0.01), ZAP70 expression (77.3% vs. 58.3%, p = 0.01), and trisomy 12 (43.5% vs. 13.7%, p < 0.001).
3.5. SPEN Mutations Carry Independent Prognostic Impact in CLL Patients
We evaluated whether SPEN mutations in CLL confer an adverse prognosis. Patients with CLL with SPEN mutations had significantly shorter time-to-first treatment compared with patients with wild type SPEN CLL (2.5 vs. 4.07 years, p = 0.01) (Figure 3). The finding of shorter time-to-first treatment in SPEN mutated CLL patients was not maintained in a multivariable analysis. IGHV unmutated status, TP53 disruption, and trisomy 12 remained independently predictive of a shorter time-to-first treatment in a multivariable analysis (Table 3).
4. Discussion
Several genes encoding proteins that regulate the NOTCH signaling pathway have been shown to be recurrently mutated in CLL. In addition to activating NOTCH1 mutations, which prolong NICD1 transcription factor activity [17], other genomic alterations have been shown to interfere with NOTCH1 signaling. These alterations include non-coding NOTCH1 mutations in the 3′ untranslated region [17], SPEN mutations [12,13,14,15,18], FBXW7 mutations [13,14,15,18,19], MED12 mutations [11,13,18], and probably alterations of the transcription factor RBPJ [12]. SPEN mutations have been described in a limited number of CLL cases in patients with treatment-naïve and relapsed/refractory disease [12,13,14,15,18]. However, the clinicopathologic features including the molecular landscape of SPEN mutations have not been extensively studied. In the current study, we assessed a large cohort of CLL patients for SPEN mutations. We identified deleterious SPEN mutations in approximately 3% of CLL cases in this cohort. The most frequent mutation co-occurring with SPEN mutation in this cohort was NOTCH1, in about half of cases. Other mutations that occurred in SPEN mutated CLL included TP53, BIRC3, SF3B1, XPO1, MUC2, FBXW7, ATM, POT1, MYD88, CXCR4, and ZMYM3.
Others have reported NOTCH1 regulatory pathway mutations in a small cohort of CLL patients [15], including five patients with SPEN mutations, and found that these patients had a significantly higher frequency of IGHV unmutated, CD38 positivity, ZAP70 positivity, deletion 11q, and trisomy 12. We also show in this cohort that SPEN mutations in CLL are significantly associated with unmutated IGHV, CD38 positivity, ZAP70 expression, and trisomy 12.
Others have shown that coding and non-coding NOTCH1 mutations negatively impact prognosis in CLL patients [17,18,20,21]. NOTCH1 activation is also associated with aggressive CLL clinical behavior and decreased time-to-first treatment [22,23], but this association does not correlate with NOTCH1 and IGHV mutational status [22]. We establish in this study that mutations of SPEN, a gene in the NOTCH1 regulatory pathway, are associated with shorter time-to-first treatment. Further studies are needed to determine whether SPEN mutations as well as other NOTCH1 regulatory pathway mutations are associated with increased transformation risk or poor response to frontline chemo-immunotherapy or targeted agents currently used for CLL patients.
NOTCH1 has a critical pathogenic role in CLL. In addition to NOTCH1 mutations, approximately half of CLL cases devoid of mutations express the active form of NOTCH1 ICN1 (intracellular portion of NOTCH1), thus implicating a much broader role for this transcription factor in CLL pathogenesis [22,23]. NOTCH1 target genes include key regulators of B-cell proliferation, survival, and signal transduction. NOTCH1 is known to transactivate MYC via binding to B-cell-specific regulatory elements, thus implicating this oncogene in CLL development. The results of this study substantially extend the role of NOTCH1 in CLL pathogenesis and have direct implications for specific therapeutic targeting.
Current International Working Group (IW) CLL guidelines recommend mutational sequencing for TP53 [24]. However, many current clinically available mutation panels sequence additional genes and provide information of undefined clinical impact. In addition, based on the adverse prognostic significance of TP53, NOTCH1, BIRC3, and SF3B1 mutations in CLL [25,26,27,28,29], Rossi et al. integrated mutations/disruptions into a cytogenetic model that independently predicts survival and improves prognostication of CLL patients [30]. We suggest that the presence of SPEN mutation may add to the currently available prognostic models.
We acknowledge the limitations of this study. Our institution is a referral center and there may be selection bias. In addition, not all patients in this cohort underwent gene mutational testing at the time of CLL diagnosis. It is therefore possible that some SPEN mutations were not present at initial diagnosis and were acquired because of clonal evolution. However, we believe that later acquisition of SPEN mutations would be unlikely as the median time from diagnosis to next generation sequencing was approximately 3 years in our cohort.
5. Conclusions
In summary, we show that SPEN mutations have a prognostic impact in CLL patients. We also show that SPEN mutations frequently co-occur with NOTCH1 mutations and are associated with similar baseline factors to NOTCH1 mutated CLL, such as IGHV unmutated status, CD38 positivity, ZAP70 expression, and trisomy 12. Most importantly, the findings we present emphasize the importance of screening for SPEN mutations as their detection may help to identify CLL patients with an adverse prognosis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chiorazzi N. Rai K.R. Ferrarini M. Chronic lymphocytic leukemia N. Engl. J. Med.200535280481510.1056/NEJ Mra 04172015728813 · doi ↗ · pubmed ↗
- 2Kipps T.J. Stevenson F.K. Wu C.J. Croce C.M. Packham G. Wierda W.G. O’Brien S. Gribben J. Rai K. Chronic lymphocytic leukaemia Nat. Rev. Dis. Prim.201731609610.1038/nrdp.2016.9628102226 PMC 5336551 · doi ↗ · pubmed ↗
- 3Rai K.R. Jain P. Chronic lymphocytic leukemia (CLL)-Then and now Am. J. Hematol.20169133034010.1002/ajh.2428226690614 · doi ↗ · pubmed ↗
- 4Paulus A. Malavasi F. Chanan-Khan A. CD 38 as a multifaceted immunotherapeutic target in CLL Leuk. Lymphoma.2022632265227510.1080/10428194.2022.209055135975791 · doi ↗ · pubmed ↗
- 5Admirand J.H. Knoblock R.J. Coombes K.R. Tam C. Schlette E.J. Wierda W.G. Ferrajoli A. O’Brien S. Keating M.J. Luthra R. Immunohistochemical detection of ZAP 70 in chronic lymphocytic leukemia predicts immunoglobulin heavy chain gene mutation status and time to progression Mod. Pathol.2010231518152310.1038/modpathol.2010.13120657554 PMC 2966512 · doi ↗ · pubmed ↗
- 6Baumann T. Delgado J. Santacruz R. Martínez-Trillos A. Royo C. Navarro A. Pinyol M. Rozman M. Poreira A. Villamor N. Chronic lymphocytic leukemia in the elderly: Clinico-biological features, outcomes, and proposal of a prognostic model Haematologica 2014991599160410.3324/haematol.2014.10732624972773 PMC 4181256 · doi ↗ · pubmed ↗
- 7Fabbri G. Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia Nat. Rev. Cancer 20161614516210.1038/nrc.2016.826911189 · doi ↗ · pubmed ↗
- 8Nadeu F. Delgado J. Royo C. Baumann T. Stankovic T. Pinyol M. Jares P. Navarro A. Martin-Garcia D. Bea S. Clinical impact of clonal and subclonal TP 53, SF 3B 1, BIRC 3, NOTCH 1, and ATM mutations in chronic lymphocytic leukemia Blood 20161272122213010.1182/blood-2015-07-65914426837699 PMC 4912011 · doi ↗ · pubmed ↗