Pleiotropic Effects on Tachyzoite and Host Cell Proteomes in Knock-Out Clones of the Open Reading Frames 297720 and 319730 Constitutively Expressed in T. gondii ShSp1 Tachyzoites
Kai Pascal Alexander Hänggeli, Joachim Müller, Manfred Heller, Anne-Christine Uldry, Sophie Braga-Lagache, David Arranz-Solís, Luis-Miguel Ortega-Mora, Andrew Hemphill

TL;DR
This study shows that knocking out specific genes in a parasite can cause widespread changes in both the parasite and host cell proteins, challenging the assumption that only the target gene is affected.
Contribution
The study reveals that gene knock-outs in T. gondii can lead to pleiotropic effects on both parasite and host proteomes.
Findings
17 common differentially expressed proteins were found in isolated tachyzoites from both knock-out clones.
76 common differentially expressed proteins were identified in host cells, linked to antiviral defense mechanisms.
KO of non-essential genes caused system-wide proteomic shifts, affecting both parasite and host gene expression.
Abstract
Toxoplasma gondii, the causative agent of toxoplasmosis widespread in animals and humans, is an intracellular apicomplexan protozoan parasite infecting a variety of host cells. Gene editing using CRISPR-Cas9 has become a standard tool to investigate the molecular genetics of this interaction. With respect to gene knock-out (KO) studies, the general paradigm implies that the gene of interest is expressed in the wildtype and that only the gene of interest is affected by the knock-out. Consequently, the observed phenotype depends on the presence or absence of genes of interest. To challenge this paradigm, we knocked out two open reading frames (ORFs) constitutively expressed in T. gondii ShSp1 tachyzoites, but not essential, namely ORF 297720 encoding a trehalose-6-phosphatase homolog and ORF 319730 encoding a You2 C2C2 zinc finger homolog. We analyzed the proteomes of tachyzoites isolated…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
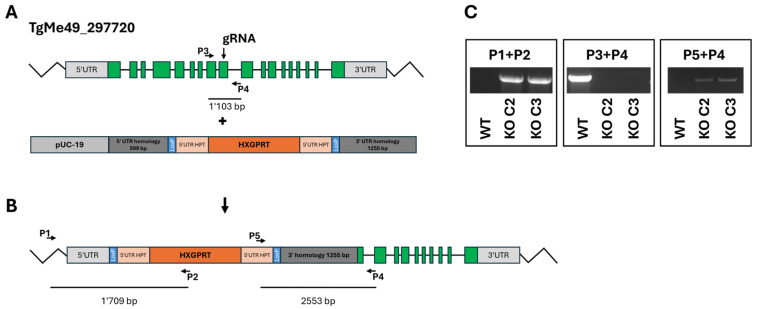 Figure 1
Figure 1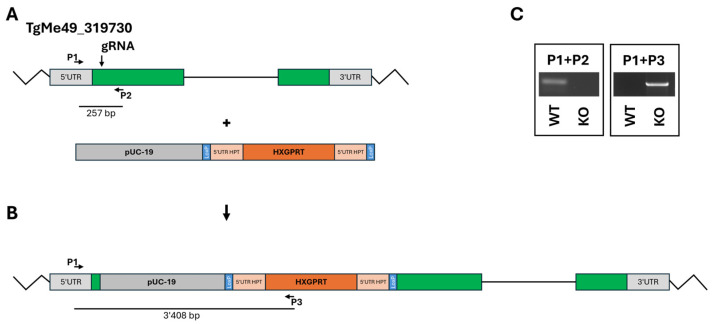 Figure 2
Figure 2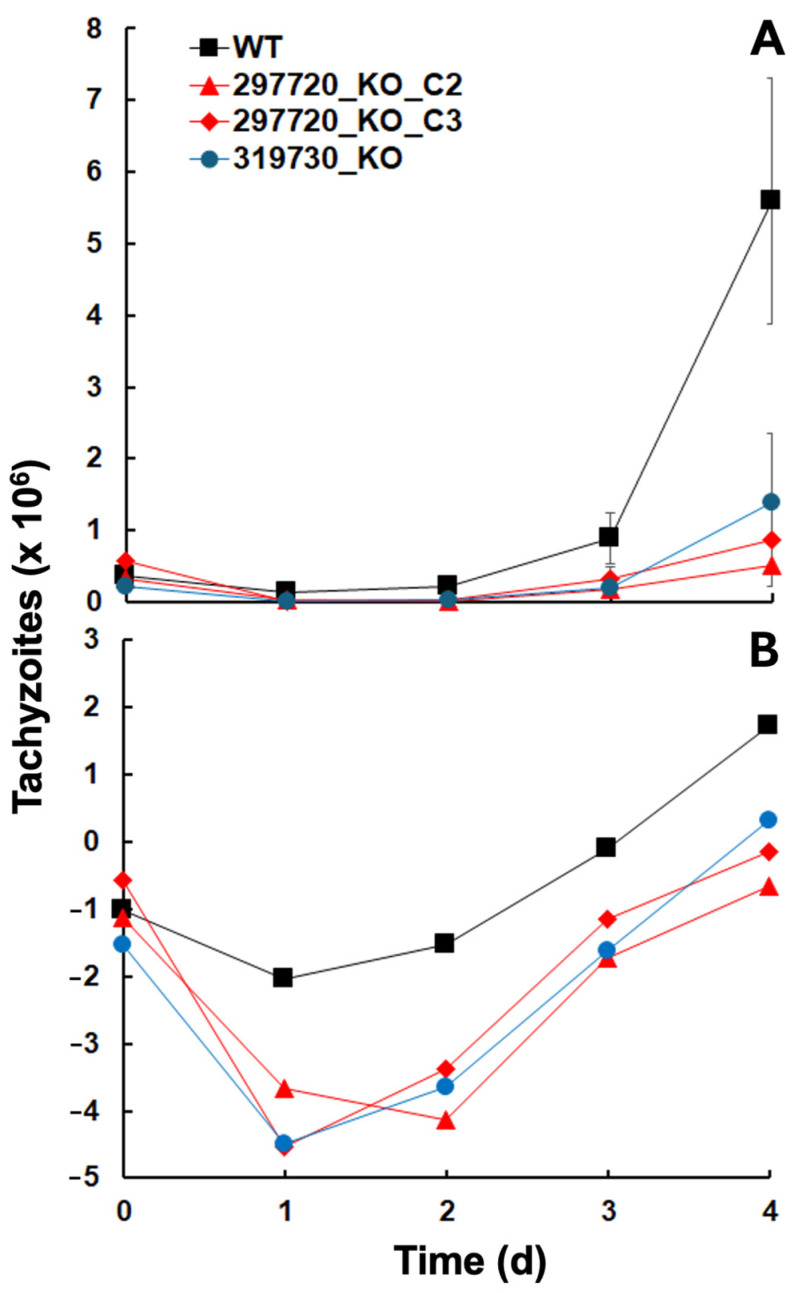 Figure 3
Figure 3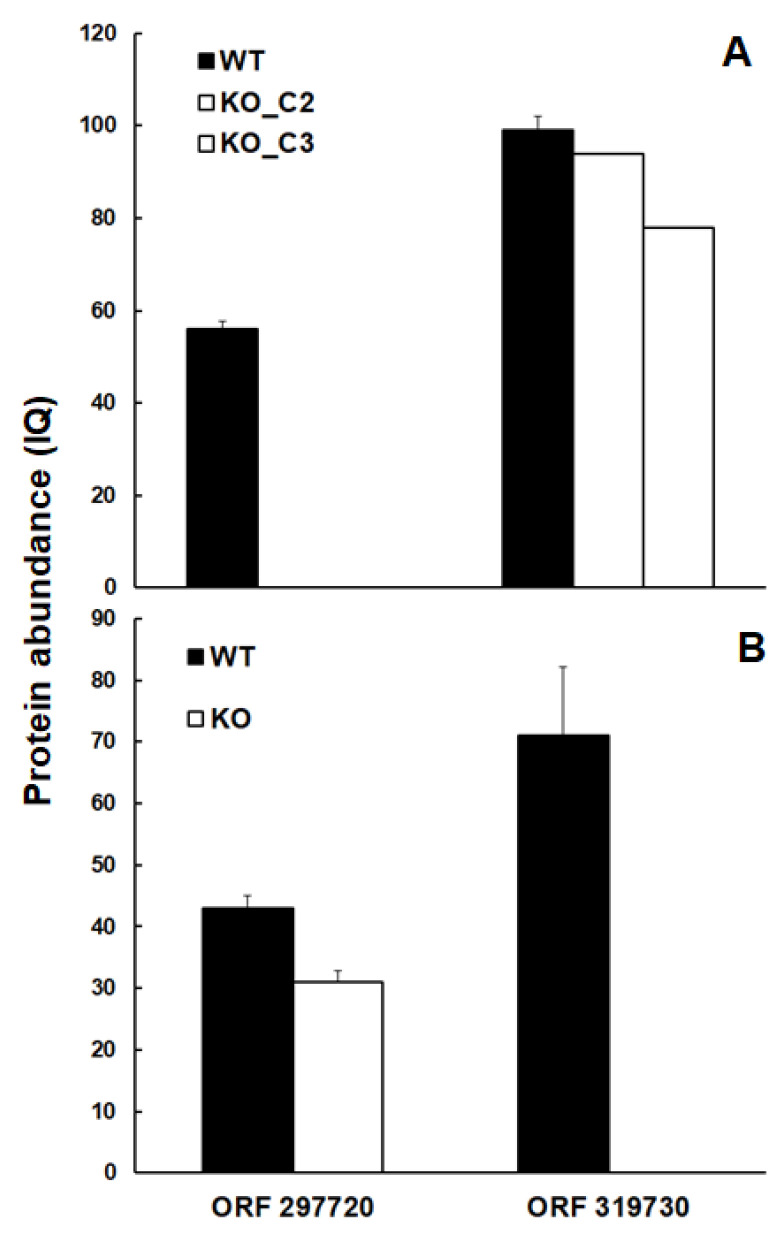 Figure 4
Figure 4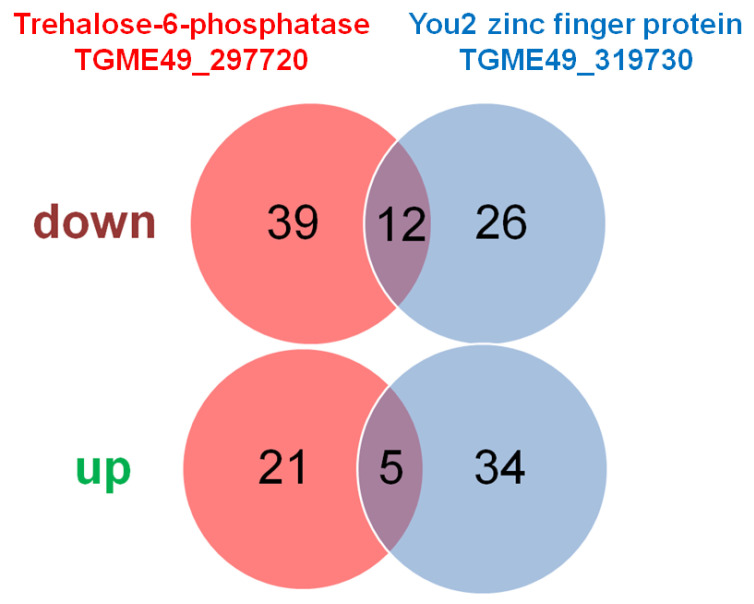 Figure 5
Figure 5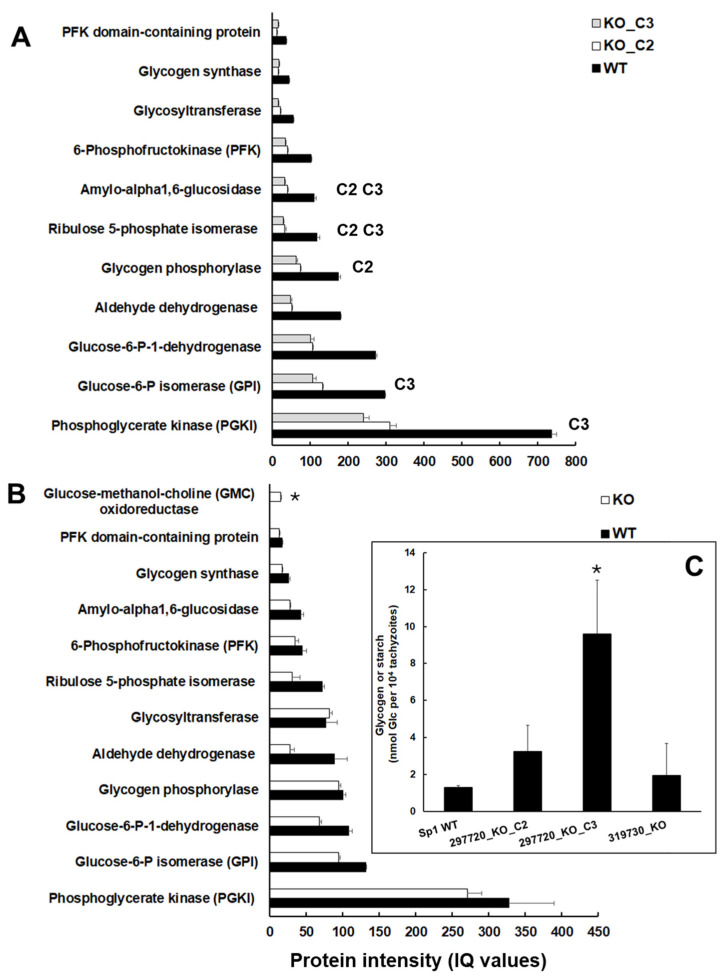 Figure 6
Figure 6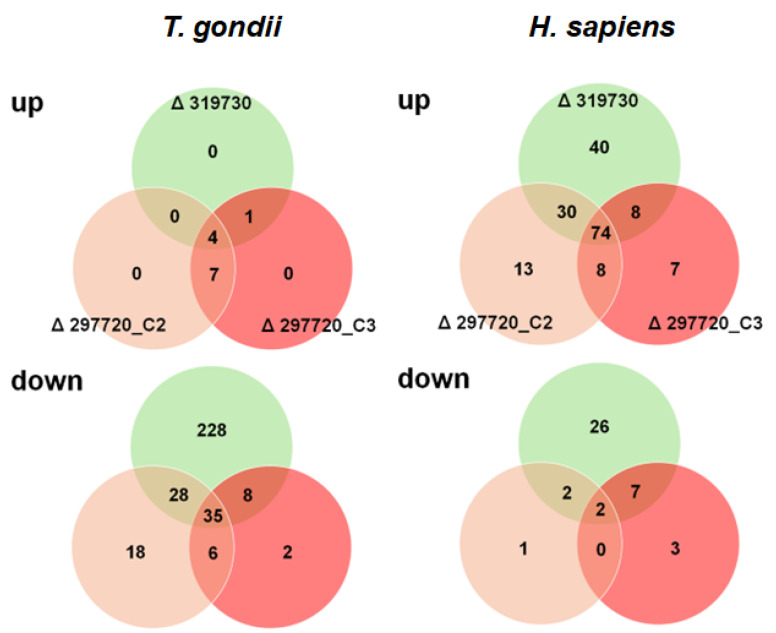 Figure 7
Figure 7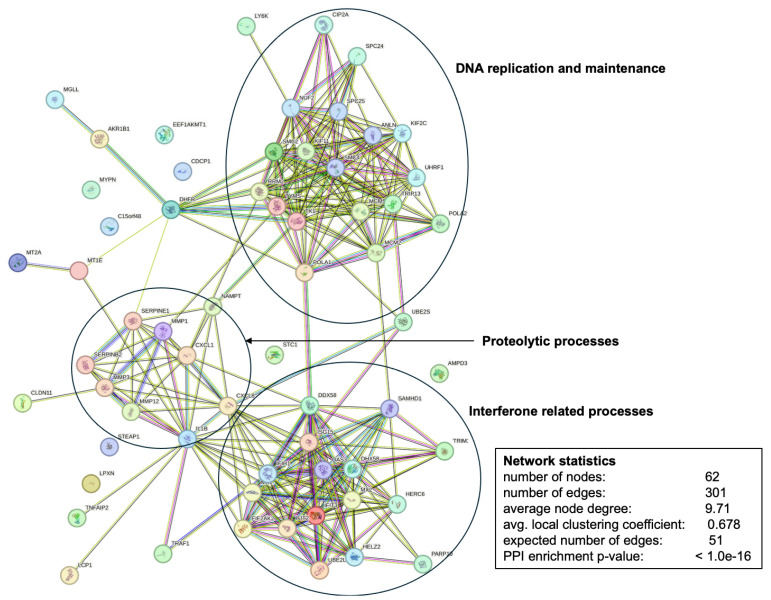 Figure 8
Figure 8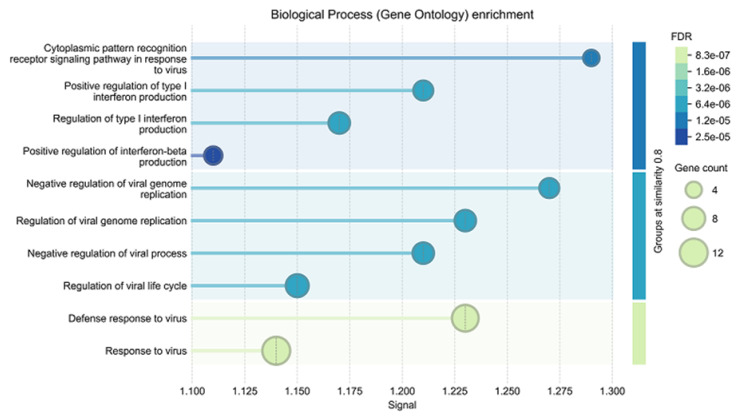 Figure 9
Figure 9- —Swiss National Science Foundation
- —Uniscientia Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsToxoplasma gondii Research Studies · Parasitic Infections and Diagnostics · Parasitic infections in humans and animals
1. Introduction
The intracellular parasite Toxoplasma gondii (Apicomplexa, Alveolata, SAR [1]) infects a broad range of host cells of animal and human origin. In humans, toxoplasmosis is one of the most prevalent parasitic diseases, with one-third of the human population on Earth being chronically infected [2,3,4]. Consequently, it is not surprising that this protozoan parasite has become a major model system to study interaction with its host, with the investigations being facilitated by its amenity to genetic manipulation. In fact, for over three decades, T. gondii has proven its excellence as a molecular genetic model [5], and the genomes of T. gondii ME49 and many other strains have been sequenced (https://toxodb.org (accessed on 4 August 2025)). With respect to genetic manipulation, gene editing via CRISPR/Cas9 has become a major tool [6,7], and genome-wide screenings have allowed us to distinguish between fitness-conferring and non-conferring genes [8].
Typical CRISPR/Cas9 studies generate knock-out (KO) or knock-in (KI) strains of selected open reading frames (ORFs) with the general hypothesis: KO of ORF X is followed by effect Y. Thus, X is functionally related to Y. However, the results obtained in KO studies can only be regarded as valid if the following prerequisites are fulfilled. (i.) The genetic modification, i.e., deletion of the ORF of interest at the correct location must be demonstrated by an appropriate method. (ii.) The transcript of the ORF of interest must be present in the corresponding wildtype and absent in the KO strains. (iii). The polypeptide encoded by the ORF of interest must be present in the wildtype but absent in the KO strains. (iv.) Unspecific effects of the genetic modification procedure must be distinguished from specific effects, e.g., by comparison with unrelated KO strains in the same genetic background and/or, if available, with different clones from the same KO population. Otherwise, a correct interpretation of the phenotypes of the KO strains cannot be realized. In most studies, points i and ii are paid attention to, whereas points iii and iv are often neglected. Despite the increasing importance of whole proteome analysis, e.g., of drug-resistant vs. susceptible strains [9], such studies on KO strains are scarce. In a previously published article, we have shown that the KO of the major surface antigen SAG1 in T. gondii RH is correlated with pleiotropic proteome changes, notably within the cell surface proteome [10]. Thus, we hypothesize that knock-out effects extend beyond targeted genes to global proteomic changes, and may trigger secondary, system-wide effects beyond the intended target. Such pleiotropic outcomes can complicate data interpretation and limit the reliability of genotype–phenotype correlations
In this study, we compared strains with KOs in two unrelated ORFs. The first ORF is TGME49_297720 encoding a 1222 amino-acid trehalose-6-phosphatase homolog expressed in tachyzoites, bradyzoites, and oocysts (https://toxodb.org; 4 August 2025). The second of which is TGME49_319730, a 149 amino-acid YOU2 C2C2 zinc finger protein homolog located in the mitochondrion (https://toxodb.org; 4 August 2025) with unknown functions and structural similarities to the human mitochondrial inner membrane protein Tim10, identified as a major binding protein of an antiprotozoal compound [11]. We investigated whether knock-out of these two ORFs leads to broader proteomic alterations in both parasite and host cells.
2. Results
2.1. Molecular Genetic Characterization of the Knock-Outs
The disruption (KO) of the ORF TgME49_297720 was intended to be obtained by double homologous recombination (Figure 1A) replacing the coding sequence with a repair template containing the hypoxanthine-guanine-phosphoribosyltransferase (HXGPRT) resistance marker (Figure 1B) in a ΔHPT background. However, PCR analysis revealed that the HXGPRT resistance cassette was inserted at the 5‘utr gRNA cutting site as shown with primers P1 + P2 (insertion of the resistance marker), P3 + P4 (disruption of TgME49_297720 at the gRNA cutting site), and P5 + P4 (insertion of the resistance cassette at the gRNA cutting site). Two clones, C2 and C3, were retained for subsequent investigations (Figure 1C).
The KO of the ORF TgME49_319730 was achieved by inserting the resistance cassette at the 5′-end of the coding sequence (Figure 2A,B). One clone with correct insertion of the resistance cassette was retained for further studies (Figure 2C).
2.2. Growth Characteristics of the KO Clones
KO clones of ORFs TgME49_297720 and 319730 could be generated, and deletions were not lethal, so the genes are not essential. However, when looking at the initial growth after inoculation of 10^5^ tachyzoites into confluent human foreskin fibroblast (HFF) cultures, it became evident that the corresponding wildtype resumed growth faster than all three KO clones (Figure 3A). A closer look at the logarithmically transformed data suggested that this was due to a decrease in the KO clone tachyzoite numbers during the first two days after inoculation (Figure 3B).
After 2 days, the growth of the KO clone tachyzoites resumed with similar growth rates as in wildtype tachyzoites (Table 1).
2.3. Differentially Expressed Proteins in Isolated Tachyzoites
In the first step, we analyzed the proteomes of isolated tachyzoites obtained from fully infected and lytic HFF cultures. Overall, 5367 non-redundant T. gondii proteins were identified in the analysis comparing two TGME_297720 knock-out strains (C2 and C3) with the corresponding wildtype T. gondii Sp1. Furthermore, 4185 proteins were identified in the analysis comparing a TGME_319730 knock-out strain with the corresponding wildtype strain (Table 2).
As shown in Figure 4, the trehalose-6-phosphate phosphatase homolog encoded by TGME49_297720 was well detectable in wildtype tachyzoites at comparative abundance levels in both experimental set-ups but was absent in the intended KO clones (Figure 4A).
The same was true for the protein encoded by TGME49_319730, which was well detectable in TGME49_297720 KO clones, but absent—as intended—in the TGME49_319730 KO clone (Figure 4B).
As expected, the proteins encoded by both ORFs subjected to KOs were absent in the respective clones. Considering the heterogeneity of the samples due to the KO, we have adapted the parameters usually applied to differential expression (DE) analysis by adding some extra filters to our scripts (Q values and PEP ≤ 0.01 at all levels and removal of low-intensity fragments). Only proteins with statistically significant differences by both ion-based quantification (IQ) and Top3 algorithms, calculated by the sum of the three most intense peptides coming from the same protein, were regarded as differentials.
Overall, 77 DE proteins were identified in tachyzoites of both TGME49_297720 KO clones vs. wildtype tachyzoites, with 51 with significantly lower abundance levels in the KO clones, and 26 with higher abundance levels in the KO clones. The comparison of TGME49_319730 KO with wildtype tachyzoites yielded 77 DE proteins. A total of 38 DE proteins had lower expression levels in KO than in wildtype tachyzoites, while 39 DE proteins had higher expression levels in KO than in wildtype tachyzoites. Twelve proteins were commonly downregulated in both KO vs. WT datasets, while five were commonly upregulated (Figure 5). The complete subsets of DE proteins in the TGME_297720 KO clones are presented in Supplemental Table S3, and the DE proteins of the TGME49_319730 KO clone is given in Supplemental Table S4.
At first, we investigated DE proteins common to both KO experiments. Within the seventeen common DE proteins, the protein family with the highest number of common DE proteins were SAG-related sequence family proteins with four differentials (one down, three up) in total, followed by four hypothetical proteins. Interestingly, the subset of common downregulated proteins contained the typical bradyzoite markers lactate dehydrogenase LDH2, and the SAG-related sequence family protein SRS35A (Table 3).
In the next step, we had a closer look at the DE proteins specific to the TGME49_297720 KO tachyzoites. We divided the 60 specific differentials (see Table S3) according to protein families and functions. Besides eighteen hypothetical proteins not annotated in any of the T. gondii strains in ToxoDB, the most abundant subset is constituted by nine SAG proteins followed by eight proteins involved in intermediary metabolism or metabolite transport (Table 4).
Of particular interest within this subset are the eight proteins involved in metabolism. As shown in Table 5, amylo-alpha-1,6-glucosidase (TGME49_226910), the key enzyme of starch degradation was downregulated in the KO tachyzoites, as well as a key enzyme of the pentose-phosphate shunt, ribulose 5-phosphate isomerase (TGME49_239310).
As expected, the metabolic proteome pattern was different in tachyzoites of the KO strain of ORF TGME49_319730 as compared to both wildtype and TGME49_297720 KO tachyzoites (Table 6).
These findings prompted us to investigate the protein intensities of glycogen/starch degrading and major glycolytic enzymes in the KO and wildtype tachyzoites in more detail. Moreover, we analyzed the glycogen/starch content in isolated tachyzoites of wildtype and KO tachyzoites. In TGME49_297720 clone tachyzoites, all enzymes were expressed at lower levels than in wildtype tachyzoites. As listed above, the levels of amyloglycosidase and ribulose-5-phosphate isomerase were significantly lower in both clones C2 and C3. Moreover, the levels of glycogen phosphorylase (TGME49_310670) were significantly lower in clone C2 only; the levels of glucose-6-phosphate isomerase (TGME49_283780) and phosphoglycerate kinase (TGME49_318230), thus of two glycolytic enzymes downstream of glycogen/starch degradation, were lower in clone C3 only (Figure 6A).
Conversely, despite tendentially lower levels, none of these enzymes had significantly lower levels in TGME49_319730 KO tachyzoites. Here, the only enzyme related to glucose metabolism with significant DE compared to wildtype was glucose-methanol-choline oxidoreductase with higher levels (Figure 6B). Consequently, it was not surprising to find higher glycogen/starch contents in clones C2 and, in particular, C3 but not in TGME49_319730 KO tachyzoites (Figure 6C).
2.4. Differentially Expressed Proteins in Infected Host Cells
In a next step, we analyzed host and parasite proteomes in infected HFF host cells. To do this, we infected confluent HFF layers and harvested the infected cells before they started to lyse. Overall, we identified 156,329 unique peptides matching 2178 T. gondii and 7755 H. sapiens proteins (Table 7 and Table S5).
With respect to T. gondii proteins, only 12 DE proteins with significantly higher levels in cells infected with KO clones vs. cells infected with wildtype tachyzoites were detected. Interestingly, the number of T. gondii proteins with lower levels in KO clone vs. wildtype infected cells was one order of magnitude higher, namely 315 in total. The comparison of TGME49_319730 KO with wildtype tachyzoites alone yielded 228 DE proteins. Conversely, in cells infected with TGME49_297720 KO clones, only 26 DE proteins with lower levels in these clones could be identified (Figure 7; Table S6).
Comparing the T. gondii DE proteins identified in isolated tachyzoites at a late stage of infection and in infected host cells at an early stage of infection, we identified twenty-seven common DE proteins (twenty upregulated and seven downregulated). Both intended KOs, namely TGME49_297720 and TGME49_319730, were within the subset of downregulated proteins, and six additional proteins were downregulated in common, including the two typical bradyzoite markers SRS35A and LDH2, and two were upregulated in common, which were the GRA proteins 80 and 82. Whereas only six common DE proteins, two downregulated and four upregulated (including three SRS proteins), were identified in the TGME49_297720_KO clones, while twelve downregulated and one upregulated protein were identified in isolated TGME49_319730_KO tachyzoites, as well as in host cells infected with this strain (Table 8).
In host cells infected with both TGME49_297720_KO strain clones, only thirteen DE proteins were identified, whereby there were seven with higher abundance, and six with lower abundance (Table 9).
Our major interest in this study were the pleotropic effects of the KO clones vs. wildtype on host cell proteomes. In fact, we could identify 218 host cell proteins with DE in cells infected with KO vs. wildtype trophozoites, whereby 38 had lower expression levels in cells infected with KO than in cells infected with wildtype tachyzoites, while 180 had higher expression levels in cells infected with KO than in cells infected with wildtype tachyzoites (Figure 7). Of major interest for the present study were the 74 proteins commonly upregulated in cells infected with KO as compared to wildtype tachyzoites. A network analysis revealed that the majority of these proteins were grouped into three clusters with functions related to DNA replication and maintenance, as well as interferon-related and proteolytic processes (Figure 8).
The complete list of host cell DE proteins is presented in Supplemental Table S7.
A gene ontology enrichment analysis of this network revealed a significant enrichment of biological processes related to antiviral responses within this network (Figure 9).
The complete list of host cell DE proteins is presented in Supplemental Table S7.
A second interaction network analysis of the 26 specific host cell proteins with higher intensities in cells infected with the TGME49_319730 KO strain as compared to cells infected with wildtype tachyzoites gave a different interaction network with a clear process enrichment in DNA replication and repair as shown in Supplemental Figure S1. In host cells infected with both TGME49_297720 KO strain clones, only eight DE host proteins (all upregulated) could be identified (Table S7).
3. Discussion
When performing knock-out (KO) or knock-in studies, the fundamental paradigm consists of attributing the observed effects in the target organism to the presence or absence of the gene of interest. This approach is valid only if there are no pleiotropic effects obliterating or masking the effects of interest. Moreover, in the case of intracellular organisms such as T. gondii (or other apicomplexans), the effects on host cells should also be considered. In the present study, we show that targeted gene knock-outs in T. gondii can lead to broader consequences than the simple loss of the gene of interest. Specifically, we observed significant shifts in the expression of both parasite and host proteomes, including sets of differentially expressed proteins that were shared across independent knock-outs.
Thus, before analyzing specific effects of the respective KOs, we examine common effects of both KOs on the tachyzoite proteomes. Concerning common downregulated proteins at early stages (i.e., within host cells) and at late stages (i.e., in isolated tachyzoites) of infection in KO vs. wildtype parasites, it is striking that the six proteins identified are upregulated in bradyzoite vs. tachyzoites (ToxoDB, accessed August 2025 and [12]). This suggests that the selection procedure following the transformation eliminates bradyzoites persisting in the untransformed wildtype from the cell population.
Only two proteins, namely GRA80 and GRA82, are upregulated in all KO vs. wildtype strains in intracellular as well as isolated tachyzoites. Both proteins are acidic and have signal peptides. Moreover, GRA80 has transmembrane domains (ToxoDB, accessed August 2025). GRA82 can be phosphorylated [13]. Both proteins are regarded as merozoite and thus cat-specific sexual stage markers were detected in infected cat intestinal tissues, and were also expressed at enhanced levels upon KOs of two transcription factors [14] and a F-box protein [15], which are in turn regarded as triggers for sexual development. Since our KO genes were different from the genes tagged in these studies, our results suggest that differential upregulation of these proteins could be caused by a pleiotropic effect due to the genetic manipulation procedure rather than specific effects due to the particular KOs.
Concerning specific effects on the KO of TGME49_297720 on tachyzoite proteomes, it is striking that—besides the intended KO—only five proteins are DE both in intracellular and purified tachyzoites, and three of them are upregulated SRS proteins. In purified KO clone tachyzoites—thus harvested at a later stage of infection—the DE proteome of this KO is larger. In particular, glycogen/starch-degrading and glycolytic enzymes are less abundant than in wildtype tachyzoites and in tachyzoites of the TGME49_319730_KO, and conversely, glycogen/starch levels are higher. This is insofar interesting as the gene product of TGME49_297720, a 1222 amino-acid protein annotated as trehalose-phosphatase, is homologous to genes having both trehalose-6-P-synthase/phosphatase domains found in insects, fungi, and plants with the highest identity to the protein CEL0385.1 of the marine algae Vitrella brassicaformis using the NCBI BLAST+ 2.17.0 server (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins, E value 0, 4 August 2025). In plants, trehalose-6-phosphate (Tre-6-P) is involved in several signaling processes including carbohydrate metabolism [16]. Similar processes may occur in plant-related apicomplexans. We have, however, no functional evidence, that the gene product of ORF TGME49_297720 encodes a functional Tre-6-P synthase and/or phosphatase. In our hands, assays with the recombinant enzyme have failed. Substrates and products may be different from canonical Tre-6-P-synthases/phosphatases. Moreover, functional activity may depend on the phosphorylation status of the protein. In fact, phosphor–proteome analyses show that the TGME49_297720 protein has multiple phosphorylation sites [13,17], which may trigger the functional activity.
Concerning the ME49_319730_KO, the number of specific DE proteins is much higher in intracellular tachyzoites (228, all downregulated) than in isolated tachyzoites (60, 26 down-, 34 upregulated). The TGME49_319730 gene product is homologous to You2 C2C2 zinc finger proteins. In mammalians, these proteins are characterized as RNA-binding proteins controlling the expression of hundreds of target RNAs [18]. If TGME49_319730 has the same function, this could explain why its absence is correlated with lower expression levels of multiple proteins at early stages of infection. However, it is unclear how its alleged mitochondrial localization fits into this picture.
The most intriguing aspect of this study is the pleiotropic effects of the genetically manipulated strains on host cells. In host cells infected with all KO strains, 74 proteins have lower expression levels than in wildtype-infected cells. Enrichment analysis suggests that these proteins are involved in antiviral defense mechanisms. This may explain the transient depression in tachyzoite numbers of these KO strains observed early after infection. It is well known that after entering a host cell, T. gondii modulates its intracellular environment by modulating host cell gene expression and consequently host cell proteomes [19]. The modulating agents are proteins secreted via rhoptries [20] and dense granules [21]. In particular, GRA16 [22] and GRA24 [21] are discussed as major effectors since both reach the host nucleus and trigger host gene expression. In our dataset, GRA24 is upregulated in host cells infected with both TGME49_297720 KO clones. However, these clones have the smallest effect on host cells in terms of host DE protein numbers. In this context, it would be worthwhile investigating to which extent GRA80 and GRA82 contribute to the modulation of host cell gene expression.
Coming back to points (i.) to (iv.) raised in the introduction, we see that, while demonstration of points i. and ii. is standard, the proteomic part of the KO strain analysis—thus points iii. and iv.—are critical and therefore most often neglected. Unambiguous identification of the proteins of interest in the wildtype and absence of the protein in the KO strain (point iii.) needs a stringent statistical approach, as shown in the present study in the case of TGME_297720. Whole-cell proteome analysis (point iv.) shows that pleiotropic effects on both parasite and host cell gene expression have to be taken into account if genes of interest are investigated in genetically manipulated strains. Consequently, appropriate controls including analysis of unrelated KO strains are needed to narrow down the spectrum of potential functions of the gene of interest. Otherwise, potential interpretation errors are programmed. However, a limitation of this study is the low number of KO clones that are investigated, and further proteomic studies on other KO strains should be carried out to substantiate these findings, ideally by also incorporating direct functional assays. Finally, non-deterministic proteome changes at the cellular level as a consequence of manipulation of a single protein mimic the unpredictable changes seen in ecosystems as a consequence of modifications of single abiotic or biotic factors [23]. This is not surprising since it is well known that chaos or non-linear responses of systems to small perturbations is common to all scales of nature [24,25,26].
4. Materials and Methods
4.1. Chemicals
If not stated otherwise, all chemical used were purchased from Sigma (St. Louis, MO, USA). Cell culture media and fetal bovine serum (FBS) were from Bioswisstec (Schaffhausen, Switzerland).
4.2. In Vitro Culture and Parasite Maintenance
Human foreskin fibroblasts (HFF, ATCC, PCS-201-101TM) were maintained in Dubecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated and filter-sterilized calf serum (FCS), and 1% Antibiotic–Antimycotic (100×) as previously described [27]. TgShSp1 and corresponding knock-out tachyzoites were cultured as previously described [28].
4.3. Isolation of Parasites
Parasites were cultured in flasks until shortly before host cell lysis. To harvest single tachyzoites, cultures were scraped and mechanically disrupted by passing the suspension three times through a 25-gauge needle. Lysed cells were centrifuged at 800× g for 10 min and washed three times with PBS, using the same centrifugation conditions. Intracellular parasites were isolated using the same procedure, but without the lysis step.
4.4. Generation of the Knock-Out Strains
A TgShSp1 strain deficient in HPT was generated at the Complutense University of Madrid, Spain following the same methodology previously applied to other strains [29]. Selection of Δhpt-tachyzoites was achieved by culturing single clones in parallel with media containing mycophenolic acid (MPA) and xanthine (25 μg/mL each), and media containing 6-thioxanthine (177 μg/mL). Parasites that proliferated in 6-thioxanthine but not in MPA-xanthine media were chosen and verified via PCR.
The single-guide RNA (gRNA) targeting the genes TgME49_319730 and TgME49_297720 were designed using the ChopChop web tool (https://chopchop.cbu.uib.no/ (accessed on 9 September 2022)). The gRNA sequences were subsequently assessed for off-target effects using a Strawberry Perl script (https://strawberryperl.com/ (accessed on 9 September 2022)) in conjunction with the latest Toxoplasma genome type 2 data base (ToxoDB-59_TgondiiME49_Genome.fasta). Primer design was carried out with Primer3Plus (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi (accessed on 9 September 2022)), with verification of GC content, hairpin formation, and potential self- and hetero-dimerization using the OligoAnalyzer Tool (https://eu.idtdna.com/calc/analyzer (accessed on 9 September 2022)). Primer sequences were then compared against known sequences using the Nucleotide BLAST+ 2.17.0 tool (https://blast.ncbi.nlm.nih.gov (accessed on 9 September 2022)) The primer sequences are presented in Supplementary Table S8.
To disrupt the gene coding for either TgME49_319730 or TgME49_297720 in the TgShSp1Δhpt background strain, a gRNA sequence targeting the gene was cloned into the pU6 vector (Addgene plasmid # 52694) using the BsaI-specific sites.
For the TgME49_319730 knock-out, this plasmid was transfected alongside the linearized pUC-HPT plasmid, which contains the HPT selection cassette flanked by two loxP sites, facilitating a potential future excision of the HPT selection cassette via Cre recombinase [30]. The transfection mixture composed of a 5.6:1 vector-insert ratio, where 28 μg of the gRNA and 2.75 μg of the selection cassette were co-transfected. 5.1e7 parasites were electroporated using a Bio-Rad GenePulser Xcell (Biorad, Cressier, Switzerland) applying a 2 mm electroporation cuvette at the following settings: 1250 V voltage, 25 μF capacitance, and infinite resistance (ꚙ Ω).
For the TgME49_297720 knock-out, the plasmid containing the gRNA was transfected alongside the linearized pUC-HPT plasmid, containing homology parts at the 3′ and 5′ site of the 5′UTR and the 3′UTR of the TgME49_297720 gene, respectively. The plasmid for the HPT selection cassette containing the homology parts was created using the primers shown in Table S8 to obtain the individual parts and the plasmid was created using the HiFi DNA Assembly Kit and the NEBuilder online tool (New England Biolabs, Ipswich, MA, USA) according to manufacturer’s protocol. The plasmid was amplified using E. coli Top10. The transfection mixture composed of a 5:1 vector-insert ratio, where 35 μg of the gRNA and 4.55 μg of the selection cassette were co-transfected. 1.3 × 10^7^ parasites were electroporated using the Nucleoflector 2b with the program U-033.
4.5. Growth Analysis
Tachyzoite growth assays were performed in 24-well plates seeded with HFF monolayers. Confluent HFF monolayers were inoculated with 10′000 tachyzoites per well. The parasites were allowed to grow until 0, 24, 48, 72, and 96 h post-infection before being harvested. At these points, the infected cell layers as well as tachyzoites in the medium supernatant were pelleted by centrifugation for 5 min at 1000× g. Subsequently, DNA extraction of the pellets was performed using the NucleoSpin Rapid Lysis Kit (Machery-Nagel, Düren, Germany) according to the manufacturer’s instructions. The extracted samples were analyzed via diagnostic PCR targeting the 529 bp repetitive fragment of T. gondii, and the number of tachyzoites per well was quantified by comparison with a tachyzoite standard series [31].
4.6. Quantification of Glycogen/Starch
Glycogen/starch was quantified in isolated tachyzoites after extraction of soluble carbohydrates as described [32,33].
4.7. Proteomic Analysis of Isolated Tachyzoites
Protein extraction and processing for mass spectrometry was performed identically as described below for the infected host cells. The mass spectrometry analysis was performed as described earlier [34]. The data was searched and quantified with Spectronaut (Biognosys) version 19.9.250422.62635 in the hybrid direct data-independent acquisition (DIA)+ (deep) mode against the ToxoDB-55 [35] T. gondii ME49-annotated proteins (with added common contaminants). Factory settings were used, including precursor qvalue cutoff = 0.01, precursor PEP cutoff = 0.2, protein qvalue cutoff (Experiment) = 0.01, protein qvalue cutoff (Run) = 0.05, and protein PEP cutoff = 0.75. Further parameters were as follows: cleavage rule = trypsin/P with maximum two missed cleavages, Carbamidomethyl (C) as fixed modification, and Acetyl (Protein N-term) and Oxidation (M) as variable modifications (maximum 5). Single hit proteins were excluded.
4.8. Proteomic Analysis of Infected Host Cells
Cell pellets were lysed in 100 μL 8 M Urea/100 mM Tris-HCl pH8, containing proteases inhibitor cocktail (Complete EDTA free, Roche, Mannheim, Germany) using 6 bursts of 10 s by a probe sonicator. Proteins were then reduced with 10 mM dithiothreitol (DTT) for 30 min at 37 C, alkylated with 50 mM Iodoacetamide for 30 min at RT in the dark, and precipitated with 5 volumes of acetone for two hours at −20 °C. Proteins were sedimented by centrifugation for 10 min at 13,000 rpm and 4 °C, and the supernatant discarded; the pellet was air dried for 15 min. Proteins were reconstituted in 30 μL 8 M Urea/50 mM Tris-HCl pH8 and protein concentration was determined by Bradford assay. An aliquot corresponding to 10 μg protein was digested for 2 h at 37 °C with sequencing grade endoproteinase LysC (Promega, Madison, WI, USA) after dilution of urea to 4 M with 20 mM Tris/HCl pH 8.0, 2 mM calcium dichloride, followed by overnight at room temperature with sequencing grade trypsin (Promega) at a urea concentration of 1.6 M. Digests were acidified with TFA (1% end concentration) and 400 ng of the digests were analyzed on a nano-liquid chromatography tandem mass spectrometer system consisting of a Vanquish Neo ultra-performace liquid chromatography (UPLC) and an Orbitrap Astral (ThermoFisher Scientific, Bermen, Germany) by loading peptides onto a C18 trap-column (PepMap 100, 5 µm, 100 Å, 300 µm i.d. ×5 mm length, ThermoFisher) at 80 bar pressure using a solvent consisting of 0.05% trifluoroacetic acid (TFA) in a water/acetonitrile mixture (98:2). Peptides were then eluted in backflush mode onto a homemade C18 CSH Waters column (1.7 μm, 130 Å, 75 μm × 20 cm) using a 22 min gradient of 5% to 40% acetonitrile in water containing 0.1% formic acid, at a flow rate of 300 nL/min. Each sample was analyzed twice, first with the data-dependent acquisition (DDA) method acquiring full scan data in the orbitrap (resolution 240′000, 380–980 m/z, normalized AGC of 300%, maximum injection time of 5 ms) and fragment spectra of peptides with charge states 2–6 in the Astral analyzer (normalized HCD energy of 28%, AGC of 80%, maximum injection time of 5 ms, scan range 120–1800 m/z, precursor exclusion for 20 s). A data-independent acquisition (DIA) mode was applied with a full scan in the orbitrap every 0.6 s (resolution 240′000, 380–980 m/z, normalized AGC of 500%, maximum injection time of 5 ms) and 299 MS2 scans of 2 m/z isolation width, with a scan range of 150–2000 m/z, and normalized AGC of 500% with a maximum injection time of 3 ms, and normalized HCD energy of 28%.
The data was searched and quantified with Spectronaut (Biognosys) version 20.1.250624.92449 in the directDIA+ (deep) mode against the ToxoDB-68 TgondiiME49-annotated proteins [35], concatenated to the UniProt [36] human sequences (release 2025_01); common contaminants were also added. Search parameters were 10 ppm and 20 ppm MS1, respectively; MS2 mass tolerance, precursor with protein q value, and PEP cutoffs were set to 0.01. Further parameters were as follows: cleavage rule = trypsin with maximum 2 missed cleavages, Carbamidomethyl (C) as fixed modification, and Acetyl (Protein N-term) and Oxidation (M) as variable modifications (maximum 3). Single hit proteins were excluded.
4.9. Statistics
Protein groups not flagged as potential contaminants were retained for further analysis. Following Pham et al. [37], the distribution of fragment peak areas reported by Spectronaut was inspected and the fragments with the lowest intensities, or not used for quantification, were removed. A further filtering step, relevant for the isolated tachyzoites datasets, consisted of enforcing the precursor and protein q value and PEP cutoffs of 0.01. Based on this selection, a leading protein was chosen per protein group on the basis of best coverage. The ion-based quantification (IQ) [37] (open-sourced-based maxLFQ) was obtained with the R package iq (version 1.10.1) for each protein group after median normalization of the fragment intensities. A Top3 [38] implementation was also provided: peptide intensities were constructed from the sum of fragment intensities and normalized by variance stabilization [39]; a same set of Top3 peptides per protein were chosen for all samples based on the sum of intensity across samples, thereby preventing minority peptides from contributing to the intensity. Moreover, proteins with less than 2 chosen peptides in a sample were deemed as not detected in this sample.
Differential expression between two groups of replicates was calculated, provided that at minimum two identifications existed in at least one group of replicates. Missing values were imputed at protein group level for the IQ measures, and at the peptide level for the Top3 measure. If there was at most one non-zero value in the replicate group for a protein group, then the missing values were imputed by drawing random values from a Gaussian distribution of width 0.3 × sample standard deviation centered at the sample distribution mean minus 2.5 × sample standard deviation at protein level, respectively, with 2.8 × sample standard deviation at peptide level. Any remaining missing values were imputed by the Maximum Likelihood Estimation (MLE) method [40]. Differential expression tests were performed with the moderated t-test of the R limma package [41]. The Benjamini and Hochberg [42] method was then applied to correct for multiple testing. The criterion for statistically significant differential expression was that the largest accepted adjusted p-value reaches 0.05 asymptotically for large absolute values of the log2 fold change, and tends to 0 as the absolute value of the log2 fold change approaches 1 (with a curve parameter of 0.1x overall standard deviation) as described recently [43]. Proteins consistently significantly differentially expressed through 20 imputation cycles for both the IQ and the Top3 measures were flagged accordingly and retained as differentially expressed.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adl S.M. Bass D. Lane C.E. Lukes J. Schoch C.L. Smirnov A. Agatha S. Berney C. Brown M.W. Burki F. Revisions to the classification, nomenclature, and diversity of eukaryotes J. Eukaryot. Microbiol.201966411910.1111/jeu.1269130257078 PMC 6492006 · doi ↗ · pubmed ↗
- 2Dubey J.P. Outbreaks of clinical toxoplasmosis in humans: Five decades of personal experience, perspectives and lessons learned Parasit. Vectors 20211426310.1186/s 13071-021-04769-434011387 PMC 8136135 · doi ↗ · pubmed ↗
- 3Beltrame A. Kim K. Weiss L.M. Human toxoplasmosis: Current advances in the field Curr. Opin. Infect. Dis.20253839240210.1097/QCO.000000000000113040747642 · doi ↗ · pubmed ↗
- 4Montoya J.G. Liesenfeld O. Toxoplasmosis Lancet 20043631965197610.1016/S 0140-6736(04)16412-X 15194258 · doi ↗ · pubmed ↗
- 5Kim K. Weiss L.M. Toxoplasma gondii: The model apicomplexan Int. J. Parasitol.20043442343210.1016/j.ijpara.2003.12.00915003501 PMC 3086386 · doi ↗ · pubmed ↗
- 6Sidik S.M. Hackett C.G. Tran F. Westwood N.J. Lourido S. Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas 9P Lo S ONE 20149 e 10045010.1371/journal.pone.010045024971596 PMC 4074098 · doi ↗ · pubmed ↗
- 7Hildebrandt F. Matias A.N. Treeck M. A CRISPR view on genetic screens in Toxoplasma gondii Curr. Opin. Microbiol.20258310257710.1016/j.mib.2024.10257739778479 · doi ↗ · pubmed ↗
- 8Sidik S.M. Huet D. Ganesan S.M. Huynh M.H. Wang T. Nasamu A.S. Thiru P. Saeij J.P. Carruthers V.B. Niles J.C. A Genome-wide CRISPR screen in Toxoplasma Identifies essential apicomplexan genes Cell 2016166142314351423–1435.e 141210.1016/j.cell.2016.08.01927594426 PMC 5017925 · doi ↗ · pubmed ↗