Challenges in the Evolving Role of Calreticulin as a Promising Target for Precision Medicine in Myeloproliferative Neoplasms
Alessandro Costa, Massimo Breccia

TL;DR
This paper reviews progress in targeting calreticulin mutations in blood cancers and highlights challenges in developing effective therapies.
Contribution
The paper outlines recent advances in immunotherapy targeting CALR mutations in MPNs and identifies key challenges for clinical translation.
Findings
Monoclonal antibodies against CALR show promising clinical responses with manageable toxicity.
Bispecific antibodies and CAR T-cell therapies offer preclinical potential against CALR-mutant cells.
Vaccination approaches have shown limited clinical benefit and remain uncertain.
Abstract
Calreticulin (CALR) mutations in myeloproliferative neoplasms (MPNs) create unique opportunities for targeted therapy. The most advanced approach involves monoclonal antibodies specifically designed to recognize the CALR neoepitope, with early clinical trials showing encouraging results. Additional preclinical data derives from bispecific antibodies that redirect T cells against CALR-mutant cells, precision antibody–drug conjugates delivering cytotoxic payloads, and CAR T-cell therapies. Vaccination against CALR-derived peptides has shown immune activity but limited benefit, and its role remains uncertain. This review discusses these advances and outlines the challenges that must be overcome to translate them into routine clinical practice. More than a decade after its discovery, advances have been made in understanding the oncogenic role of mutant CALR in BCR::ABL1-negative…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
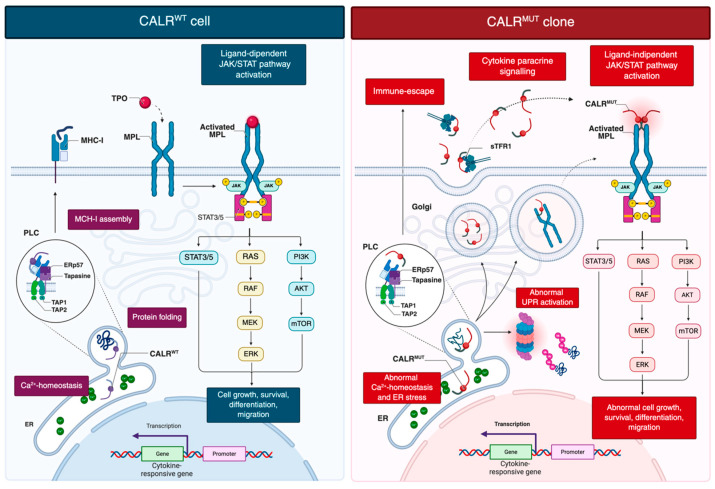 Figure 1
Figure 1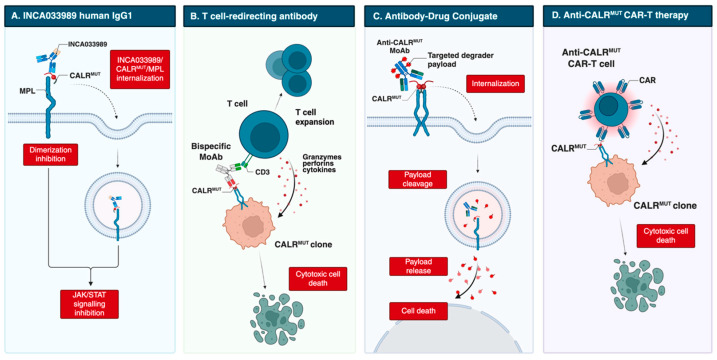 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMyeloproliferative Neoplasms: Diagnosis and Treatment · Acute Myeloid Leukemia Research · Chronic Myeloid Leukemia Treatments
1. Introduction
BCR::ABL1-negative myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are clonal hematopoietic stem cell disorders characterized by sustained myeloid proliferation and a variable risk of thrombosis, hemorrhage, and fibrotic or leukemic progression [1]. Their clonal origin was first suggested in the 1970s [2], but major advances in understanding occurred with the identification in 2005 of the JAK2 V617F mutation, present in nearly all PV cases and in a majority of ET and PMF. Soon after, activating mutations in MPL, the thrombopoietin (TPO) receptor, were identified in a smaller subset of ET and PMF [3]. A further breakthrough came in 2013 with the identification of somatic insertions and deletions in exon 9 of the CALR gene, which encodes calreticulin, occurring in 20–25% of ET and PMF patients without JAK2 or MPL mutations [4,5].
In recent years, interest in CALR mutation has steadily grown, shifting from their initial diagnostic and prognostic relevance to their potential as direct therapeutic targets. The unique biochemical features of mutant CALR, including its interaction with MPL and its immunogenic neoepitope, have opened new avenues for targeted therapies. Considering these advances, we examine the evolving significance of CALR in MPNs together with open issues for integrating immune-based strategies into the management of CALR-positive diseases.
2. Genetic and Molecular Pathways in CALR-Mutated MPNs
2.1. Structure and Function of Calreticulin
Wild-type CALR is a conserved endoplasmic reticulum (ER) lumen chaperone essential for intracellular calcium (Ca^2+^) homeostasis, protein quality control, and glycoprotein folding (Figure 1) [6].
The CALR gene comprises nine exons and encodes a protein organized into three functional domains: an N-terminal lectin-like domain that binds glycans, a central proline-rich (P) domain that interacts with polypeptides, and a C-terminal domain ending with the ER-retention signal KDEL (Lys-Asp-Glu-Leu) [7].
Functionally, the C-terminal domain binds Ca^2+^ with low affinity, thereby contributing to ER calcium buffering [8]. The KDEL sequence ensures retention of CALR within the ER, where it facilitates the proper folding of N-glycosylated proteins destined for the plasma membrane, extracellular space, or other cellular compartments [9]. As a holdase chaperone, CALR preferentially binds glycosylated precursors through its lectin domain, preventing aggregation, premature oligomerization, oxidation, and degradation of misfolded proteins [10].
CALR also plays a key role as an extracellular “eat-me” signal expressed on the surface of stressed or apoptotic cells. Once externalized, CALR binds to phosphatidylserine and functions as a damage-associated molecular pattern (DAMP), thereby promoting macrophage-mediated phagocytosis [11]. The P domain of CALR has been proposed to serve as a molecular bridge to phagocytic receptors, including low-density lipoprotein receptor-related protein 1 (LRP1) and C1q, enhancing its pro-phagocytic function [12]. In parallel, activated macrophages have been shown to secrete CALR, which binds to viable target cells and marks them for clearance through a mechanism of programmed cell removal (PrCR) [13].
2.2. Advances in Molecular Mechanisms and Oncogenic Transformation of CALR-Mutated MPNs
Since their discovery in 2013, more than fifty distinct CALR mutations have been identified. All are located within exon 9 and produce a +1 base pair (bp) frameshift [5]. Based on structural features and the extent of acidic domain loss, they are categorized as type 1 mutations (52-bp deletion; CALR^Del52^), with complete loss of the acidic C-terminal region, or type 2 mutations (5-bp insertion; CALR^Ins5^), with partial retention of the acidic domain [4,5]. The remaining 20% are designated as type 1-like or type 2-like mostly according to structural predictions based on α-helical content. Mutants with higher predicted α-helical content are considered type 2-like, whereas those with lower content resemble type 1 [14,15,16].
Structural studies have provided insight into the abnormal interaction between mutant CALR and MPL. Neither the intrinsic chaperone activity of CALR nor its polypeptide-binding domain is necessary for MPL activation. Instead, three features are critical: specific N-glycosylation sites on MPL, the lectin-binding ability of mutant CALR, and the positively charged residues in its novel C-terminal tail generated by the frameshift [17,18,19,20]. Replacing these basic residues with neutral glycine, or partially deleting the mutant tail, reduces the CALR transforming capacity without affecting MPL binding, indicating that receptor binding and activation are distinct processes [21].
Mutations in CALR have shown to affect key aspects of cellular biology and immune regulation (Figure 1) [22,23,24]. Loss of the ER-retention KDEL sequence directs mutant CALR into the secretory pathway, enabling pathological MPL activation [4,5]. The altered C-terminal tail triggers an N-domain conformational change, exposing the N-glycan binding pocket for recognition of immature N-glycans on MPL [25]. Direct electrostatic interactions are also thought to occur between the positively charged residues in the mutant C-terminal domain and negatively charged residues within the D1 domain of MPL [25]. This interaction, occurring in the ER and Golgi, facilitates trafficking of the CALR–MPL complex to the cell surface, where mutant CALR acts as a cytokine mimic, promoting ligand-independent MPL dimerization and sustained JAK/STAT activation [19,26]. Additional data indicate that homomultimerization of mutant CALR, driven by its aberrant C-terminal sequence, is necessary for full receptor activation [25,27]. Mutant CALR was also detected in plasma of MPN patients [28]. Pecquet et al. [28] investigated its biological role and demonstrated a correlation between soluble mutant CALR (smCALR) levels and mutational burden. They further showed that the prolonged half-life of smCALR results from its complex formation with soluble transferrin receptor 1 (sTFR1). Functionally, smCALR probably binds to and activates MPL on megakaryocyte progenitor cells, promoting ligand-independent proliferation and sensitizing neighboring cells to CALR-mediated signaling.
2.3. Clonal Dynamics and Open Challenges
Over the past decade, preclinical models and genomic analyses have provided critical insights into clonal evolution, tracing the trajectory of hematopoietic stem cells (HSCs) from early subclinical expansion to overt disease [29]. In particular, investigations into self-renewal dynamics and somatic mutation–based lineage tracing have shown that JAK2 V617F mutations may arise decades before diagnosis [30], and in some cases even in utero [31]. A striking parallel has been reported for CALR in a unique case involving monozygotic twins who developed CALR-mutated MF in adulthood. Both individuals harbored the same somatic CALR mutation, likely acquired in one twin and transmitted to the other through transplacental hematopoietic cell sharing. Subsequent clonal divergence was evidenced by the presence of a TET2 mutation in only one twin [32]. Mathematical modeling studies suggest that, unlike JAK2, CALR mutations may arise later in life but confers a stronger proliferative advantage, thereby accelerating clonal expansion [33]. Interestingly, some evidence showed that the proportion of mutated cells is not equally distributed within the progenitors, with CALR-mutated HSCs biased towards myeloid differentiation and CALR-mutated cells heavily enriched in megakaryocyte progenitors [23,34].
However, translational research on CALR remains limited by several constraints inherent to current models, including species-specific differences, reliance on ectopic overexpression, and limited phenotypic penetrance [35]. Transgenic mouse models, including Calr^Del52^;Calr+/+ and heterozygous knock-in lines, typically develop thrombocytosis without progression to fibrosis [36,37]. Homozygous models exhibit more pronounced features, such as marked thrombocytosis, reduced bone marrow erythropoiesis, splenomegaly, and extramedullary hematopoiesis, but lack a competitive repopulating advantage in transplantation assays [36,38,39]. More recently, it has also been shown that HSCs with CALR haploinsufficiency display MPN-like features, including reduced bone marrow erythropoiesis and splenic extramedullary hematopoiesis; the combination of CALR^Del52^ and CALR haploinsufficiency restores the self-renewal capacity of mutant HSCs, eliminating the competition from wild-type cells and aiding the expansion of mutant clones towards MPN [40].
Recently, CRISPR/Cas9 editing has enabled the targeted introduction of CALR mutations directly into the endogenous mouse locus, generating in-frame deletions of 19, 52, or 61 bp. However, these models also exhibit only mild thrombocytosis and limited JAK/STAT activation [41,42]. To overcome these limitations, a novel humanized knock-in model was developed in 2023, in which common CALR mutations were introduced into healthy human HSCs using CRISPR/Cas9 [35]. Consistent with earlier findings, CALR-mutated HSCs did not show immediate proliferative advantage, suggesting that additional transcriptional or microenvironmental events are required to drive transformation. Interestingly, some mice transplanted with human CALR-mutated HSCs developed marrow fibrosis and splenomegaly, potentially reflecting a greater sensitivity of human MPL to TPO compared with its murine counterpart [35]. These findings support the concept that CALR-driven transformation is not cell-autonomous but instead depends on a complex interplay between the driver mutation, intrinsic transcriptional programs, and extrinsic environmental signals. Nonetheless, further analyses are needed to fully understand the oncogenic dynamics of CALR and to overcome the limitations of current models.
3. Immunotherapeutic Strategies in CALR-Mutated MPNs
3.1. Anti-CALR Monoclonal Antibodies
The advent of immunotherapy has transformed the treatment landscape of hematologic malignancies. Among these, monoclonal antibodies (MoAbs) occupy a central role due to their ability to selectively target malignant cells, limiting off-target toxicity and enabling durable responses even in relapsed or refractory disease [43]. While well established in lymphoproliferative disorders and acute leukemias, their application in MPNs is more recent, yet they represent the most advanced immunotherapeutic approach in both preclinical and early clinical development (Table 1).
The cell-surface localization of mutant CALR makes it an attractive target. The first preclinical evidence, reported in 2020, described B3, a murine chimeric MoAb recognizing the mutant CALR C-terminal domain [44]. In CALR^Del52^ ET murine model, B3 reduced bone marrow megakaryocyte counts and improved thrombocytosis. Subsequently, Achyutuni et al. [38] treated homozygous CALR^Del52^ transgenic mice with a murine IgG2a antibody directed against the human CALR neoantigen, achieving a rapid decline in platelet counts after five doses over 2.5 days, with rebound to baseline within 24 h after discontinuation.
In 2022, two independent groups advanced this concept. Mughal et al. [45] generated antibodies targeting internal peptides within the mutant CALR C-terminal, defining key epitope. In parallel, Tvorogov et al. [46] developed 4D7, an IgG2α MoAb specific for a C-terminal sequence common to type 1 and type 2 CALR mutations. 4D7 selectively disrupted the mutant CALR-MPL interaction, attenuated hyperactivation of the JAK/STAT pathway, suppressed thrombopoietin-independent megakaryocytic proliferation, and inhibited differentiation of CALR-mutated CD34+ cells, prolonging survival in xenograft models, including ruxolitinib-resistant disease.
However, these antibodies were generated in non-human species and had incompletely characterized mechanisms of action. A major advance came with INCA033989, developed by Reis et al. [47]. This fully human IgG1 MoAb harbors an N297A Fc mutation that abolishes Fc-mediated effector functions (Figure 2). INCA033989 binds recombinant CALR^Del52^ (K_d_ 1.75 nM) and CALR^Ins5^ (K_d_ 6.78 nM), with preferential binding to CALR^Del52^, likely reflecting epitope exposure differences in the CALR^Del52^-MPL complex. The MoAb/CALR/MPL complex relies on dynamin-dependent endocytosis; whether mutant CALR or antibody binding affects MPL recycling remains unclear. Functionally, INCA033989 inhibited pSTAT3/pSTAT5 in CALR-mutated MF CD34+ cells in a dose-dependent manner, without affecting wild-type, JAK2-mutated, or MPL-mutated cells. In a CALR-mutated ET murine model, it induced hematologic and molecular responses by selectively targeting mutant hematopoietic stem cells while sparing normal hematopoiesis. In a CALR^Del52^/TP53 Q317 double-mutant post-MPN AML xenograft, it selectively targeted mutant cells and prolonged survival [48].
To date, two phase 1 clinical trials are underway for INCA033989, specifically the INCA33989-101 (NCT05936359, ex-US) and INCA3989-192 (NCT06034002, US). Both are multicenter, open-label, first-in-human studies with a phase 1a dose-escalation stage in high-risk MF or ET patients harboring exon 9 CALR mutations, followed by phase 1b expansion and phase 1c crossover with ruxolitinib in MF patients with suboptimal response. Preliminary data in high-risk ET (n = 49) treated with doses ranging from 24 mg to 2500 mg administered intravenously every two weeks show encouraging activity without dose-limiting toxicities. As per protocol design, hydroxyurea (HU) or anagrelide as cytoreductive therapy was allowed, while prohibited medications included interferon, thalidomide, busulfan or lenalidomide, requiring a washout period of 5 half-live prior to study enrollment. A rapid reduction in platelet count was reported, with hematologic responses achieved within 4 weeks, an overall response rate (ORR) of 79%, and complete responses (CR) in 66% of patients. Reductions in variant allele frequency (VAF) were observed in 88% of patients, with 25% achieving a ≥50% reduction after 12 weeks of treatment. Most adverse events were grade 1–2, with transient, asymptomatic grade 3 events mainly consisting of lipase elevations [49]. Early dose-dependent normalization of platelet counts and reductions in clonal burden were observed; definitive results are awaited.
Additional antibodies are designed to recruit immune effector cells, mediating antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC). In 2023, Kuchnio et al. [50] reported JNJ-88549968, a bispecific T-cell–redirecting antibody selectively targeting MPN clones. This agent induced specific binding and T-cell activation against CALR-mutated cells in vitro, including autologous assays, and in xenograft models, regardless of mutation subtype; it is now in a phase 1 trial (NCT06150157).
Another T-cell–redirecting antibody, INCA035784, is a fully human IgG1 with a silenced Fc domain. Unlike INCA033989, it binds an N-domain region of mutant CALR conserved across C-terminal mutations [51]. It does not recognize wild-type CALR exposed on the cell surface after doxorubicin-induced stress, and binding is unaffected by soluble CALR. In vitro, it induces dose-dependent T-cell activation and cytotoxicity against CALR-mutated CD34+ cells from both type 1 and type 2 cases.
Finally, precision antibody–drug conjugates (pADCs) have been explored to deliver cytotoxic payloads selectively to malignant cells. One approach couples degraders of SMARCA2/4 or CDK9 to an anti-CALR MoAb, leveraging dysregulation of the SWI/SNF chromatin-remodeling complex, which governs gene expression programs essential for hematopoietic stem cell maintenance [52,53,54]. Dual degradation disrupts aberrant SWI/SNF function, inducing cytotoxicity in progenitors and restoring normal hematopoiesis. Preclinical data show selective binding to mutant cells, inhibition of megakaryocytic growth and differentiation in patient-derived CALR-mutated samples, and durable disease regression. Further studies are ongoing [52].
3.2. Vaccination Strategies Against Exon 9 CALR Mutations and Immune Checkpoint Inhibition
Therapeutic cancer vaccines aim to elicit immune responses directly against tumor-specific antigens [55]. In MPNs, exon 9 CALR mutations generate unique neoantigens, making them attractive targets. Early studies showed that patient-derived T cells could recognize and eliminate autologous CALR-mutated cells ex vivo, but their activity was weaker than that of healthy donor T cells stimulated with the same peptides [56,57]. To optimize immunogenicity, Gigoux et al. [58] used in silico prediction to identify mutant CALR neoepitopes with strong MHC-I binding; however, these alleles were underrepresented in CALR-mutated compared to JAK2-mutated cases, potentially restricting vaccine applicability.
Clinically, the CALRlong36 peptide vaccine was evaluated in the phase 1 trial NCT03566446 [59]. Ten CALR-mutated MPN patients received 15 doses over one year. Ex vivo T-cell responses emerged in 8 patients, yet no hematologic or molecular responses were achieved. Attempts to enhance efficacy through combinatorial approaches have so far been unsuccessful. In NCT05444530, dual CALR/JAK2 vaccination was combined with the CTLA-4 inhibitor ipilimumab in 14 patients [60]. Mutant CALR-specific immune responses were observed in 35.7%, but no reduction in allele burden occurred, leading to early trial discontinuation.
3.3. CAR T-Cell Therapy Targeting Mutant CALR
Preclinical efforts have also extended to CAR T-cell therapy. Schueller et al. [61] showed that murine CALR-mutant-specific CAR-T cells could target Ba/F3-hMPL cells expressing human CALR^Del52^ and prolong survival in xenografts, although no remissions were achieved in immunocompetent chimeric mice. More recently, Rampotas et al. [62] reported a CALR-directed CAR T-cell strategy capable of selectively eradicating CALR-mutated human cell lines, irrespective of expression levels. In NGS xenografts harboring CALR-mutated AML cells, this approach reduced leukemic burden and improved survival. In ex vivo assays using CD34+ cells from MPN patients, depletion ranged from 40% to 90% with limited off-target toxicity toward JAK2 V617F samples.
4. Targeting CALR in the Era of Precision Medicine: Issues to Be Addressed
From the earliest observations it became clear that CALR-mutated MPNs display distinctive clinical features compared with other driver mutations. Indeed, CALR-mutated patients, as first reported by Klampfl and Nangalia [4,5], are younger and characteristically show marked thrombocytosis accompanied by relatively lower hemoglobin and leukocyte counts when compared with JAK2-mutated cases. In ET, CALR mutations further delineate a subgroup with reduced thrombotic risk [63]. While survival in ET appears independent of driver mutation status [64,65], type 1 CALR mutations in MF are consistently associated with improved outcomes [66,67,68]. These findings have informed the evolution of prognostic scoring systems [69]. It is important to note, however, that CALR attenuate but do not eliminate the adverse prognostic impact of high molecular risk (HMR) mutations and unfavorable karyotype [70]. These clinical and prognostic insights raise key considerations for the development of therapies targeting CALR-mutated clones.
4.1. Will Anti-CALR Therapies Shift Management from Thrombotic Risk Reduction to Disease Modification?
An open question is how anti-CALR therapies might reshape current management strategies and in which clinical settings their use would be most impactful. The potential to modify the natural history of the disease by directly targeting the mutant clone raises the issue of whether therapeutic goals should move beyond thrombotic risk reduction. This is especially relevant in ET, where current management is primarily focused on reducing thrombotic risk. While HU remains the standard treatment for high-risk IPSET-thrombosis patients and intermediate-risk cases requiring cytoreduction, unmet therapeutic needs persist. Anagrelide can be considered in patients who are resistant or intolerant to HU [71], although clinically significant anemia and long-term bone marrow fibrosis, particularly in CALR-mutated cases, have raised concerns [72,73]. Busulfan and pipobroman are effective alternatives but carry potential leukemogenic risks [74]. In addition, ruxolitinib did not demonstrate superiority over HU in the MAJIC-ET trial [75]. By contrast, pegylated interferon-α (Peg-IFNα) has shown efficacy in controlling thrombocytosis, reducing spleen size, and improving symptom burden [76,77], with notable molecular responses, particularly in JAK2 V617F–mutated cases. Similar results have recently been reported for ropeginterferon α-2b (Ropeg-IFN) in patients with PV, and full results from ongoing studies in ET, including the SURPASS-ET trial, are eagerly awaited [78].
Similar issues arise in CALR-mutated MF, where management largely follows the principles applied to other driver mutations, yet distinct clinical considerations warrant attention. Pivotal trials of ruxolitinib and fedratinib have only partially addressed whether driver mutation status influences outcomes [79]. In a retrospective analysis of 29 CALR-mutated MF patients in the COMFORT-II trial, 20 received ruxolitinib and demonstrated safety, efficacy, and survival comparable to the overall cohort [80]. More recently, momelotinib was approved for MF with anemia due to its dual JAK1/JAK2 inhibition and blockade of activin A receptor type 1 (ACVR1). Among 79 JAK inhibitor-naïve patients treated with momelotinib, 14% carried type 1 CALR and 3% type 2 CALR mutations [81], with type 1 CALR patients showing significantly higher 3-, 5-, and 10-year survival even after multivariate adjustment for transplantation. Real-world data highlight additional nuances. In a cohort of 1.055 MF patients treated with ruxolitinib, 135 with CALR mutations more frequently developed anemia and increased blast percentages [82], likely reflecting later treatment initiation, higher disease burden, and longer intervals from diagnosis, which may diminish the favorable prognostic impact of CALR mutations.
Allogeneic hematopoietic stem cell transplantation (HSCT) remains the only curative option. Advances in technique and prognostic tools have broadened its feasibility. In 2023, Hernández-Boluda et al. [83] reported outcomes in 346 CALR-mutated MF patients undergoing HSCT, with five-year survival of 63% versus 50% in JAK2-mutated cases, though no distinction was made by CALR subtype or HMR status. More recently, Gagelmann et al. [84] analyzed clonal dynamics in 324 patients after reduced-intensity conditioning: 23% were CALR-mutated, achieving molecular clearance in 73% at day 30 and 82% at day 100, compared with 42% and 63% for JAK2-mutated cases and 54% and 100% for MPL-mutated cases, potentially explaining the superior post-transplant outcomes in CALR- and MPL-mutated patients.
Taken together, anti-CALR therapies represent an important opportunity to achieve disease modification, something rarely attainable with current approaches. As ongoing studies mature, they will clarify whether these agents can meaningfully reduce thrombotic risk, delay disease progression, and improve quality of life in patients with ET and MF.
4.2. Are We Ready for Measurable Residual Disease Monitoring?
The prospect of targeted therapy capable of eradicating the mutant clone highlights the importance of molecular monitoring, akin to measurable residual disease (MRD), to evaluate treatment efficacy over time and guide clinical decisions. Improvements in blood counts, symptoms, or organ involvement do not always correlate with molecular responses [85].
Although current management of PV and ET focuses largely on thrombo-hemorrhagic risk reduction, preventing fibrotic or leukemic progression remains a critical research goal. This is supported by evidence linking disease progression to increasing allele burden during follow-up [86]. While the JAK2 V617F VAF exhibits considerable interindividual variability in MPNs, CALR-mutated cases show a more homogeneous distribution, typically between 40% and 50% [87,88,89]. In patients with CALR VAF ≤20%, lower platelet and neutrophil counts and improved overall survival have been reported, independent of mutation subtype, age, or thrombotic history [88]. Conversely, higher allele burdens are associated with increased acquisition of additional mutations, greater risk of anemia during follow-up, and more frequent progression to fibrosis [87,89].
To date, no therapy has convincingly altered the natural history of MPNs. As previously said, IFNα has demonstrated not only complete hematologic responses but also molecular responses, outcomes not achieved with cytoreduction using anagrelide or HU [90]. However, in CALR-mutated patients, clonal reduction dynamics are variable even among molecular responders [91], and the reduction of the CALR-mutant clone with Peg-IFNα is generally less pronounced than in JAK2 V617F-mutated patients [92,93]. The MAJIC-PV trial provided evidence that molecular responses in PV patients treated with ruxolitinib correlate with improved overall and progression-free survival [94]. In addition, Guglielmelli et al. [85] prospectively measured changes in JAK2 and CALR VAF in 77 PV or ET patients treated with ruxolitinib, reporting reductions from a median of 68% to 3.5% in JAK2-mutated cases and from 49% to 4% in a CALR-mutated ET patient. Molecular responses were also observed in three CALR-mutated ET patients treated with ruxolitinib in the MAJIC-ET study [75].
Several techniques have been evaluated for monitoring allele burden, including fragment length analysis (FLA), Sanger sequencing, and next-generation sequencing (NGS) [86,91]. Jones et al. [95] compared detection limits of CALR assays using cell lines with 61 bp deletions, reporting detection limits of 10–25%, 5%, 5%, and 1.2% for Sanger sequencing, FLA, high-resolution melt analysis, and NGS, respectively. While FLA is rapid and cost-effective for diagnostic screening, it has a relatively high detection limit and is unsuitable for precise quantification of fragments of different sizes, often overestimating shorter fragments [91,96]. Similarly, Sanger sequencing, with 10–20% sensitivity, is inadequate for MRD monitoring [97]. NGS is highly sensitive but limited by cost and turnaround time, particularly for allele burdens below 1–5%. Quantitative PCR (qPCR) offers higher sensitivity and broad availability, but its application to CALR is challenged by the complex assay design required due to nucleotide repeats in exon 9. This approach has been employed to quantify type 1 and type 2 mutations, with a detection limit that generally does not exceed 0.1%, remaining suboptimal for reliable MRD monitoring [97].
In contrast, digital droplet PCR (ddPCR) overcomes the limitations of conventional PCR by partitioning the reaction into thousands of microdroplets, each analyzed individually. The low DNA copy number per droplet minimizes competition between primers, enabling highly accurate quantification of allele burden [84,86,91]. Using this approach, mutant CALR alleles can be detected down to 0.01%, providing a highly sensitive and precise tool for MRD assessment [96,98]. A clear example of its clinical utility is the monitoring of MRD post-HSCT in myelofibrosis. Early studies demonstrated that CALR mutations are cleared more rapidly and frequently post-transplant compared with MPL or JAK2 V617F mutations [99]. Patients with detectable mutations at days +100 and +180 had a significantly higher risk of relapse, irrespective of the underlying driver mutation. More recent evidence indicates that mutation clearance as early as day +30 is a strong predictor of both relapse and post-transplant survival, emphasizing the importance of timely molecular monitoring [84].
Nevertheless, defining molecular response as a marker of disease modification requires careful standardization and validation of threshold levels, which must ultimately translate into meaningful clinical benefit. Importantly, clearance of the dominant mutant clone does not necessarily equate to clearance of all clones, highlighting the need for further studies to clarify the clinical relevance of clonal monitoring in MPNs.
4.3. How to Face the Immune Escape Issue?
Accumulating evidence supports the presence of an immunosuppressive environment in CALR-mutated MPNs. While the underlying mechanisms remain incompletely understood, they appear primarily related to impaired antigen presentation and immunosuppressive remodeling of the tumor microenvironment. Impaired phagocytosis of tumor cells has been observed in CALR^Del52^ knock-in mice, consistent with an immunosuppressive role of CALR-mutated clones; this defect was associated with reduced T-cell activation and expansion of regulatory T cells (Tregs) [100], rather than affecting phagocytic capacity of macrophages [101]. Additional studies have shown that CALR^Del52^ variant drives the expansion of TGF-β1-producing erythroid progenitors and Tregs, while suppressing T-cell cytotoxicity [102].
Moreover, although CALR mutations can elicit T-cell responses, CALR-mutant-specific responses are attenuated in MPN patients compared with healthy individuals [103]. High expression of immune checkpoint receptors such as PD-1 and CTLA-4, consistent with T-cell exhaustion, has been reported. Furthermore, CALR-mutant–specific T cells from patients are predominantly CD4+, and the absence of CALR-mutant-specific CD8+ T cells may reflect impaired antigen presentation [103]. Under normal conditions, CALR participates in the peptide-loading complex (PLC) along with ERp57 and tapasin, ensuring proper assembly and high-affinity peptide loading onto major histocompatibility complex (MHC) class I molecules [104]. Proteasome-derived peptides are transported into the endoplasmic reticulum by the TAP1/TAP2 heterodimer, trimmed by ERAP1, and loaded onto MHC class I heavy chains and β2-microglobulin with the assistance of tapasin, ERp57, and CALR. Fully assembled complexes are then exported via the Golgi to the cell surface to mediate immune recognition of abnormal cells [105]. By guiding MHC class I molecules to the PLC and stabilizing peptide loading, CALR is essential for antigen presentation. Mutant CALR disrupts this process, impairs high-affinity peptide binding, and reduces MHC class I expression at the cell surface, likely facilitating immune evasion of CALR-mutated clones [106].
Collectively, these findings highlight immune escape as a major barrier in CALR-mutated MPNs, likely contributing to the limited success of T-cell-based and vaccination strategies. Continued efforts to overcome this hurdle will be essential for the development of effective therapies.
5. Conclusions
CALR mutations and their downstream pathophysiologic effects offer unique opportunities for precision targeting. Despite highly encouraging preclinical results demonstrating selectivity and low toxicity, challenges persist. While the biological rationale and preclinical evidence strongly support the development of mutant CALR-directed therapies, clinical success will require overcoming disease-intrinsic immune evasion, refining patient selection, and implementing robust strategies for clonal burden monitoring.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arber D.A. Orazi A. Hasserjian R.P. Borowitz M.J. Calvo K.R. Kvasnicka H.-M. Wang S.A. Bagg A. Barbui T. Branford S. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data Blood 20221401200122810.1182/blood.202201585035767897 PMC 9479031 · doi ↗ · pubmed ↗
- 2Adamson J.W. Fialkow P.J. Murphy S. Prchal J.F. Steinmann L. Polycythemia Vera: Stem-Cell and Probable Clonal Origin of the Disease N. Engl. J. Med.197629591391610.1056/NEJM 197610212951702967201 · doi ↗ · pubmed ↗
- 3Pikman Y. Lee B.H. Mercher T. Mc Dowell E. Ebert B.L. Gozo M. Cuker A. Wernig G. Moore S. Galinsky I. MPLW 515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia P Lo S Med.20063 e 27010.1371/journal.pmed.003027016834459 PMC 1502153 · doi ↗ · pubmed ↗
- 4Nangalia J. Massie C.E. Baxter E.J. Nice F.L. Gundem G. Wedge D.C. Avezov E. Li J. Kollmann K. Kent D.G. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK 2N. Engl. J. Med.20133692391240510.1056/NEJ Moa 131254224325359 PMC 3966280 · doi ↗ · pubmed ↗
- 5Klampfl T. Gisslinger H. Harutyunyan A.S. Nivarthi H. Rumi E. Milosevic J.D. Them N.C.C. Berg T. Gisslinger B. Pietra D. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms N. Engl. J. Med.20133692379239010.1056/NEJ Moa 131134724325356 · doi ↗ · pubmed ↗
- 6Michalak M. Groenendyk J. Szabo E. Gold L.I. Opas M. Calreticulin, a Multi-Process Calcium-Buffering Chaperone of the Endoplasmic Reticulum Biochem. J.200941765166610.1042/BJ 2008184719133842 · doi ↗ · pubmed ↗
- 7Houen G. Højrup P. Ciplys E. Gaboriaud C. Slibinskas R. Structural Analysis of Calreticulin, an Endoplasmic Reticulum-Resident Molecular Chaperone Cellular Biology of the Endoplasmic Reticulum Agellon L.B. Michalak M. Springer International Publishing Cham, Switzerland 20211325978-3-030-67696-410.1007/978-3-030-67696-4_234050860 · doi ↗ · pubmed ↗
- 8Wijeyesakere S.J. Gafni A.A. Raghavan M. Calreticulin Is a Thermostable Protein with Distinct Structural Responses to Different Divalent Cation Environments J. Biol. Chem.20112868771878510.1074/jbc.M 110.16919321177861 PMC 3058961 · doi ↗ · pubmed ↗