Emergence of Livestock‐Associated Methicillin‐Resistant Staphylococcus aureus ST398 in Wild Birds, Brazil
Mateus Rocha Ribas, Felipe Vásquez‐Ponce, Rodrigo Cardoso, Dany Mesa, Gustavo Rocha, Victor Felipe Wolleck, Juliana Lemos Dal Pizzol, Izadora Borgmann Frizzo de Assunção, Vinicius Pais e Oliveira, Gabriel Salvador, Amanda Tfardoski Rodrigues, Gregory Batista Melocco

TL;DR
Wild birds in a Brazilian forest were found to carry antibiotic-resistant MRSA ST398, likely originating from livestock, showing how human activities can spread microbes to wildlife.
Contribution
First report of MRSA ST398 in Brazilian wildlife, linking livestock-associated strains to wild birds in a protected area.
Findings
12.2% of wild birds carried S. aureus, including two MRSA ST398 isolates.
MRSA ST398 strains showed broad antimicrobial resistance and virulence genes.
Phylogenomic analysis linked MRSA ST398 to swine in northeastern Brazil.
Abstract
Antimicrobial‐resistant and virulent Staphylococcus aureus strains are spreading across diverse environments and hosts, but studies on Brazilian wildlife remain limited. From April to December 2021, oropharyngeal swabs were collected from 197 wild birds spanning five orders, 25 families, and 54 species in São Camilo State Park, a protected Atlantic Forest fragment facing significant pressure from surrounding agricultural landscapes. S. aureus was detected in 12.2% of the birds, including 27 methicillin‐susceptible S. aureus (MSSA) and two Methicillin‐resistant (MRSA) isolates. MSSA strains showed high inducible Macrolide‐Lincosamide‐Streptogramin B (MLSB) resistance, with 37% carrying the blaZ gene for penicillin resistance, and antimicrobial‐resistant isolates frequently harboring the scn gene. Genomic sequencing identified both MRSA strains as ST398, marking the first report of MRSA…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
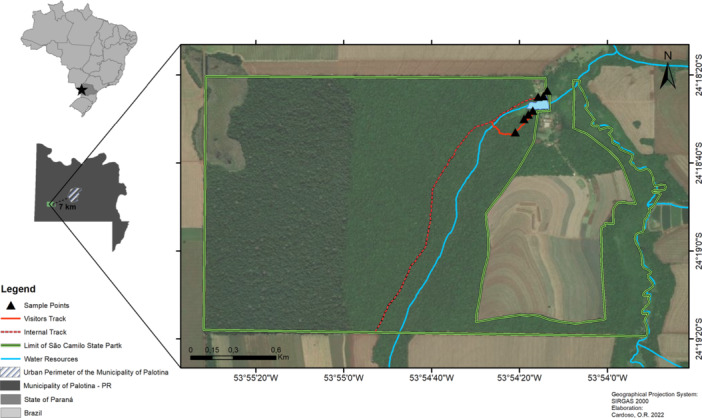 Figure 1
Figure 1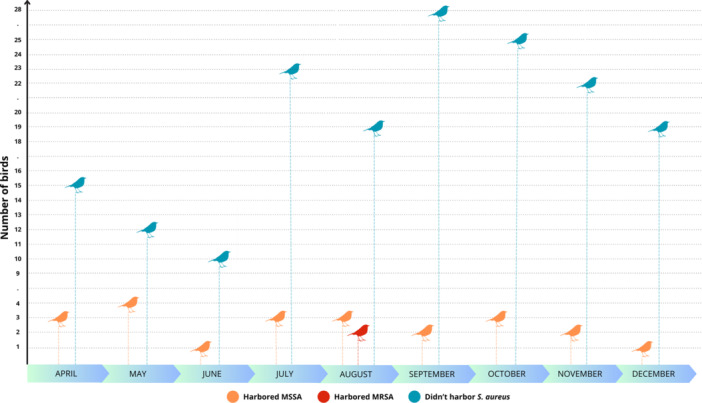 Figure 2
Figure 2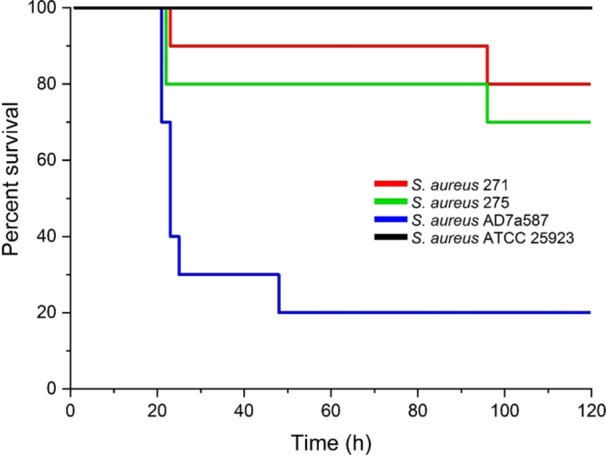 Figure 3
Figure 3 Figure 4
Figure 4| ID | Specie | Order | Family | Isolate | Resistance phenotype | Resistance genes | Virulence genes |
|---|---|---|---|---|---|---|---|
| 14 |
| Passeriformes | Thraupidae | 46 | Susceptible | (‐) |
|
| 15 |
| Passeriformes | Corvidae | 48 | CLI ‐ ERI ‐ GEN ‐ AMI ‐ TOB | (‐) |
|
| 16 |
| Passeriformes | Parulidae | 53 | CLI ‐ ERI | (‐) |
|
| 54 | CLI ‐ ERI | (‐) |
| ||||
| 29 |
| Passeriformes | Pipridae | 107 | PEN ‐ CLI ‐ ERI ‐ TOB |
|
|
| 30 |
| Passeriformes | Thraupidae | 108 | PEN ‐ CLI ‐ ERI |
|
|
| 31 |
| Passeriformes | Parulidae | 109 | CLI ‐ ERI | (‐) |
|
| 32 |
| Passeriformes | Parulidae | 111 | PEN ‐ CLI ‐ ERI |
|
|
| 41 |
| Passeriformes | Passerellidae | 135 | PEN ‐ CLI ‐ ERI |
|
|
| 55 |
| Passeriformes | Parulidae | 172 | PEN ‐ CLI ‐ ERI |
|
|
| 62 |
| Columbiformes | Columbidae | 191 | Susceptible | (‐) |
|
| 192 | Susceptible | (‐) |
| ||||
| 67 |
| Cuculiformes | Cuculidae | 208 | Susceptible | (‐) |
|
| 72 |
| Passeriformes | Thraupidae | 221 | PEN ‐ CLI ‐ ERI |
|
|
| 75 |
| Piciformes | Picidae | 229 | PEN ‐ CLI ‐ ERI |
|
|
| 79 |
| Passeriformes | Turdidae | 240 | AMI ‐ TOB | (‐) |
|
| 120 |
| Passeriformes | Pipridae | 324 | Susceptible | (‐) |
|
| 333 | Susceptible | (‐) |
| ||||
| 121 |
| Passeriformes | Thamnophilidae | 325 | CLI ‐ ERI | (‐) |
|
| 326 | CLI ‐ ERI | (‐) |
| ||||
| 138 |
| Passeriformes | Thraupidae | 364 | CLI ‐ ERI | (‐) |
|
| 151 |
| Columbiformes | Columbidae | 395 | PEN ‐ CLI ‐ ERI |
|
|
| 153 |
| Passeriformes | Tityridae | 399 | PEN ‐ CLI ‐ ERI |
|
|
| 162 |
| Passeriformes | Icteridae | 425 | CLI ‐ ERI | (‐) |
|
| 426 | CLI ‐ ERI | (‐) |
| ||||
| 176 |
| Passeriformes | Tyrannidae | 462 | PEN ‐ CLI ‐ ERI |
|
|
| 191 |
| Passeriformes | Rhynchocyclidae | 496 | Susceptible | (‐) |
|
| Isolate Information |
|
|
|---|---|---|
| Host |
|
|
| Taxonomic Group | Order: Passeriformes ‐ Family: Cardinalidae | Order: Passeriformes ‐ Family: Pipridae |
| Source | Oropharynx | Oropharynx |
| Antibiotic Resistance Phenotype | PEN ‐ FOX ‐ CLI ‐ ERY ‐ CIP ‐ NOR ‐ LEV ‐ GEN ‐ TOB ‐ TET ‐ SUL | PEN ‐ FOX ‐ CLI ‐ ERY ‐ CIP ‐ LEV ‐ GEN ‐ TOB ‐ TET ‐ SUL |
|
| ||
| Genome size (bp) | 2,761,481 | 2,761,987 |
| Contig number | 61 | 61 |
| CDSs | 2,508 | 2,510 |
| N. rRNAs | 1 | 1 |
| N. tRNAs | 14 | 14 |
| % GC | 32.7% | 32.7% |
|
| ||
| MLST | ST398 | ST398 |
| SCC | SCC | SCC |
|
| t1456 | t1451 |
|
| ||
| Aminoglycosides |
|
|
| β‐lactams |
|
|
| Macrolides |
|
|
| Fluoroquinolones |
|
|
| Phenicols |
|
|
| Lincosamides |
|
|
| Tetracycline |
|
|
| Trimethoprim |
|
|
| Efflux pumps |
|
|
|
| ||
| Exoenzymes |
|
|
| Toxins |
|
|
| Clumping factor |
|
|
| Hemoglobin receptor |
|
|
| Adhesin (PIA) synthesis |
|
|
| Serine‐aspartate repeat Sdr proteins |
|
|
| Capsular polysaccharide synthesis |
|
|
| Type VII secretion system |
|
|
| others |
|
|
- —Coordenação de Aperfeiçoamento de Pessoal de Nível Superior Financing Code 001.
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Resistance in Staphylococcus · Salmonella and Campylobacter epidemiology · Bacterial biofilms and quorum sensing
Introduction
1
Antimicrobial resistance is one of the greatest threats to global health, with Staphylococcus aureus emerging as a key pathogen in this crisis due to its remarkable adaptability and ability to colonize diverse hosts and thrive across diverse environments (Haag et al. 2019; Silva et al. 2020; Howden et al. 2023; Song et al. 2024). This versatility allows it to exist as a commensal organism in the microbiota of humans and animals while also posing significant threats by causing infections ranging from mild to life‐threatening (Krismer et al. 2017; Guo et al. 2020; Cheung et al. 2021). The rise of antimicrobial resistance has propelled S. aureus to the forefront of One Health research, particularly regarding methicillin‐resistant S. aureus (MRSA), which is resistant to nearly all beta‐lactams and often exhibits high virulence and multidrug resistance (Lee et al. 2018; Algammal et al. 2020).
Classified as a high‐priority pathogen by the World Health Organization (World Health Organization. 2024), MRSA has extended its presence beyond hospital and community settings into livestock and, more recently, into natural ecosystems (Lakhundi and Zhang 2018; Hou et al. 2023). This expansion highlights the growing interconnectedness between human, animal, and environmental health (Denissen et al. 2022). Furthermore, resistant and virulent MRSA strains have been detected in wild animals, including mammals and birds (Heaton et al. 2020; Abdullahi et al. 2021; Martínez‐Seijas et al. 2023), raising concerns about their potential spillover into natural ecosystems and wildlife populations (Dolejská 2020; Ramos et al. 2022; Ramos and Cunha 2024).
Brazil, with its vast territorial expanse, is home to an exceptional diversity of biomes, making it one of the most biodiverse countries on the planet. Among these, the Atlantic Forest stands out as a biodiversity hotspot, rich in species yet severely threatened by urban expansion and the conversion of land into agricultural and livestock areas (Rezende et al. 2018; Grelle et al. 2021; Casallanovo et al. 2024). This transformation of the landscape fosters closer interactions among humans, domestic animals, and wildlife, thereby heightening the risk of pathogen spillover events and accelerating the spread of antimicrobial‐resistant bacteria into natural and protected environments (Faust et al. 2018; Gibb et al. 2020; Wiemeyer et al. 2021; Blanco‐Peña et al. 2024; Ribas et al. 2025).
Birds, as crucial sentinels and reservoirs of antimicrobial‐resistant bacteria (Bonnedahl and Järhult 2014), are especially relevant in this context. However, studies on the microbiota of free‐living wildlife in Brazil, particularly regarding S. aureus, remain scarce, limiting the understanding of how environmental changes like forest fragmentation influence pathogen dynamics from a One Health perspective.
This study aims to address this gap by investigating S. aureus strains in wild birds inhabiting a protected fragment of the Atlantic Forest, an area under significant agricultural pressure. The research focuses on analyzing their resistance patterns and virulence traits, shedding light on the potential risks these strains pose to the ecosystem and public health. Notably, for the first time, we report MRSA isolates of the international ST398 clone in wild birds in Brazil, highlighting the ecological implications of the dissemination of high‐priority pathogens within Atlantic Forest fragments. Also, the increasing presence of livestock‐associated strains in Brazilian natural environments.
Materials and Methods
2
Study Settings and Sample Collection of Wild Birds
2.1
This study was conducted in São Camilo State Park (hereafter “São Camilo”), a 385.34‐hectare protected area of Seasonal Semideciduous Forest within the Atlantic Forest biome, located in the municipality of Palotina, western Paraná, southern Brazil (24°18′20″ S, 53°54′15″ W) (Figure 1). The park is subject to intense anthropogenic pressure due to surrounding agro‐industrial expansion, leading to significant habitat fragmentation. To investigate the presence of antimicrobial‐resistant Staphylococcus aureus in wild bird populations, seven mist nets were installed at distinct locations within the park and operated over a 28‐day period between April and December 2021. Nets were deployed at dawn and remained active for 6 h daily. Captured birds (n = 197) across five orders, 25 families, and 54 species (Appendix 1) were carefully removed, placed in cotton fabric holding bags, and transported to a field station for sample collection and species identification. At the station, oropharyngeal swabs were collected from each bird and immediately immersed in Brain Heart Infusion (BHI) broth supplemented with 6.5% NaCl for incubation. Following sampling, all birds were released at their capture sites. The migratory behavior of the sampled bird species was classified following the criteria established by Somenzari et al. (2018).
Location of São Camilo State Park, situated in the municipality of Palotina, state of Paraná, Brazil, highlighting the mist nets (black arrows) placed throughout the study.
Culture Processing, Bacterial Identification, and Phenotypic and Genotypic Characterization
2.2
Samples were incubated at 37°C for 18 to 24 h and then cultured on 5% sheep blood agar plates. Isolate identification was conducted using Matrix‐Assisted Laser Desorption/Ionization Time‐of‐Flight (MALDI‐TOF) mass spectrometry (Bruker MALDI Biotyper). Antimicrobial susceptibility testing was performed by disk‐diffusion method against the following antimicrobial agents (in µg/disk): penicillin G (10 U), cefoxitin (30), clindamycin (2), erythromycin (15), gentamicin (10), amikacin (30), tobramycin (10), ciprofloxacin (5), levofloxacin (5), norfloxacin (10), tetracycline (30), tigecycline (15) and trimethoprim/sulfamethoxazole (1.25/23.75). The double‐disk diffusion method with clindamycin and erythromycin disks was performed to determine Macrolide‐Lincosamide‐Streptogramin B (MLSB) resistance phenotypes. Brazilian Committee on Antimicrobial Susceptibility Testing—BrCAST 2025 guidelines were followed, except for testing with penicillin G, which followed the Clinical and Laboratory Standards Institute—CLSI. 2025 standards. The reference strains S. aureus ATCC 25923 and S. aureus ATCC 29213 were used as quality control strains. On the other hand, the antimicrobial resistance genes mecA, blaZ, ermA, ermB, ermC, ermT, and aacA‐aphD, along with virulence genes sea, seg, seh, sen, tst, lukED, lukFS, icaAD, icaBC, sasG, fnbpA, fnbpB, and scn, were selected for genotypic characterization of the isolates by polymerase chain reaction (PCR). Multiplex PCR was conducted to determine the SCCmec type in methicillin‐resistant isolates. To control for DNA extraction quality, a partial region of the 16S rDNA gene was also amplified by PCR. The used primers and amplification conditions are detailed in Appendix 2.
In Vivo Virulence Assays in the Galleria mellonella Infection Model
2.3
To evaluate the virulence potential of MRSA strains, an in vivo experiment was carried out with the Galleria mellonella infection model (Tsai et al. 2016; Sheehan et al. 2019). Groups of 10 larvae (230–280 mg) were inoculated with 10 μL of 2 × 10^6^ CFU/larvae of each bacterial strain in PBS. A control group was injected with 10 μL PBS to rule out death from physical trauma. S. aureus AD7a587 was used as a virulent control, while S. aureus ATCC 25923 served as a non‐virulent control. After treatment, larvae were incubated at 37°C, and survival was monitored hourly for 120 h. Two biological replicates were done per strain. Survival curves were plotted with the Kaplan‐Meier method, and the log‐rank test (p < 0.05) was used for statistical analysis (Origin Software, USA).
Whole Genome Sequence Analysis
2.4
The genome sequencing of the samples confirmed that MRSA was performed. For the whole genome sequence, genomic DNA was extracted using the PureLink™ Genomic DNA Mini Kit (Thermo Fisher Scientific, USA). DNA quality and concentration were assessed by NanoDrop spectrophotometer (Thermo Scientific) and Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA), respectively. The library was constructed using the Nextera DNA Prep kit (Illumina, San Diego, CA, USA) and sequenced using the NextSeq. 550 paired reads platform (2 × 75 bp). Reads were trimmed with Trimmomatic and assembled using CLC Genomics Workbench 12.0.3. The read quality was verified using FastQC (Andrews 2010) version 0.12.1. These reads were subjected to the genomic assembly process using SPAdes (Bankevich et al. 2012) version 3.15.5 (options –cov‐cutoff 30 and –phred‐offset 33). Contigs smaller than 500 base pairs were filtered out. For the identification of virulence and resistance genes in MRSA strains, the ABRicate program (Torsten, n.d.) was used. Within ABRicate, the genomes were analyzed using the following databases: NCBI AMRFinderPlus (Feldgarden et al. 2019), CARD (Jia et al. 2016), ARG‐ANNOT (Gupta et al. 2014), Resfinder (Zankari et al. 2012), MEGARES 2.0 (Doster et al. 2020), PlasmidFinder (Carattoli et al. 2014), and VFDB (Chen et al. 2016). To identify spa tandem repeats, the spa region was analyzed online using spaTyper (http://spatyper.fortinbras.us/), and results were cross‐verified with the Ridom spa Server (https://www.spaserver.ridom.de/) for accurate classification. For SCCmec detection, in silico PCR was performed with Geneious Prime (2024.0.4; Biomatters), both to confirm conventional PCR findings and to screen for a broader range of SCCmec types (I to XI). This analysis used gene targets proposed by Yamaguchi et al. (2020).
Phylogenetic Analysis
2.5
For phylogenetic analysis based on the core‐genome, 1661 strains of Staphylococcus aureus belonging to ST398 circulating at the human‐animal‐environment interface were obtained from NCBI and Pathogenwatch (Argimón et al. 2021) (https://pathogen.watch/genomes/all?genusId=1279&mlst=398&speciesId=1280). All genomes used (1663) were annotated using the Prokka program (Seemann 2014) version 1.14.6, and the annotation files in GFF format were submitted to the Panaroo program (Tonkin‐Hill et al. 2020) using MAFFT (Katoh and Standley 2013) as the aligner (options ‐a core ‐‐aligner mafft) to obtain the already aligned core‐genome (file in ALN extension). Once the aligned core‐genome was obtained, the file was submitted to the IQ‐TREE program (von Haeseler et al. 2014) using the ultrafast bootstrap method (Hoang et al. 2018) with 1000 replicates and the Tamura‐Nei model (Tamura and Nei 1993) (options ‐B 1000 ‐m TN93) to generate the phylogenetic tree of all analyzed strains. Once the preceding phylogenetic relationship has been established, a comparative cgMLST analysis of the 21 closest genomes from S. aureus circulating at the human–animal interface was constructed using a concatenated filtered core‐gene alignment from 2335 genes (2,060,121 bp in length) obtained with Panaroo software (https://github.com/gtonkinhill/panaroo), and FastTree 2.1.11. The SNP‐sites software (https://github.com/sanger-pathogens/snp-sites) was used to obtain Single Nucleotide Polymorphisms (SNPs), and the snp‐dists 0.8.2 software (https://github.com/tseemann/snp-dists) was used to build the SNP matrix. Tree visualization, country, host, MRSA. Antimicrobial susceptibility and heatmap of antibiotic resistome were performed using iTol v.6 (https://itol.embl.de/).
Genomic Sequencing Repository
2.6
All assembled genomes of the MRSA strains from this study have been deposited in NCBI with the following identifiers: JBAGCW000000000 for the strain hosted by Cyanoloxia glaucocaerulea (isolate 271 ‐ SQ688) and JBAGCX000000000 for the strain hosted by Manacus manacus (isolate 275 ‐ SQ696).
Results
3
Characterization of Methicillin‐Susceptible Staphylococcus aureus
3.1
S. aureus was detected in 12.2% of the sampled birds, resulting in 29 isolates from 24 individuals across four orders (Passeriformes, Columbiformes, Piciformes, and Cuculiformes), 15 families, and 20 species. Of these, 17 species exhibit non‐migratory behavior (i.e., are resident), while only three are known to display partial migratory patterns (Table 1; Appendix 1). The isolates were recovered in every month of the study (Figure 2). Twenty‐seven isolates were identified as Methicillin‐Susceptible Staphylococcus aureus (MSSA). From those, approximately 70% (n = 19) exhibited inducible MLSB resistance, although none harbored the evaluated erm genes. Around 37% (n = 10) of the MSSA isolates carried the blaZ gene, conferring resistance to penicillin, and 11% (n = 3) of these isolates showed resistance to at least one tested aminoglycoside, despite the absence of the aacA‐aphD gene. Seven isolates were susceptible to all tested antimicrobial agents (Table 1).
Monthly sampling of wild birds in São Camilo State Park, southern Brazil (April–December 2021), focusing on the temporal distribution of Staphylococcus aureus isolates detected in the sampled birds.
Regarding the virulence gene profile, six strains carried the lukED gene, and one isolate of these isolates also harbored the seh enterotoxin gene. The scn gene was present in 74% (n = 20) of MSSA isolates. Notably, only one isolate carried both lukED and scn genes. The chromosomal intercellular adhesion gene cluster icaABCD, which is associated with biofilm production, was found in all isolates. Additionally, the fibronectin‐binding protein A gene was present in all isolates, while the fibronectin‐binding protein B gene was absent. The surface‐anchored protein G gene was identified in five isolates (Table 1).
Characterization of Methicillin‐Resistant Staphylococcus aureus
3.2
Two MRSA isolates were found in Cyanoloxia glaucocaerulea and Manacus manacus, both exhibiting a broad resistance phenotype. The MRSA isolate from C. glaucocaerulea was resistant to penicillin, cefoxitin, ciprofloxacin, norfloxacin, levofloxacin, gentamicin, tobramycin, tetracycline, and trimethoprim/sulfamethoxazole, along with constitutive MLSB resistance. Similarly, the isolate from M. manacus showed resistance to penicillin, cefoxitin, ciprofloxacin, levofloxacin, gentamicin, tobramycin, tetracycline, and trimethoprim/sulfamethoxazole, and exhibited constitutive MLSB resistance to clindamycin and erythromycin. Due to the high‐priority status of MRSA as a pathogen and its broad antimicrobial resistance, both isolates were selected for comprehensive genome analysis.
Genomic and Phylogenetic Characterization of MRSA Isolates
3.3
Genomic analysis identified both MRSA isolates as belonging to sequence type (ST) 398, a lineage often associated with livestock worldwide. The isolate from C. glaucocaerulea was classified as spa type t1456 (tandem repeat sequence: 08‐16‐02‐25), while the isolate from M. manacus was classified as spa type t1451 (tandem repeat sequence: 08‐16‐02‐25‐34‐25). The SCCmec element in both isolates was confirmed as type V via conventional and in silico PCR.
An extensive resistome was found in both MRSA strains, with resistance genes identified for multiple antimicrobial classes. Detected genes included those conferring resistance to β‐lactams (mecA, blaZ), aminoglycosides [ant(6)‐Ia, aac(6′)‐aph(2″), spw, ant(9), aph‐Stph, aac(3)], macrolides (ermT), phenicols (fexA), lincosamides (isaE, inuB), tetracycline (tetM, tetL), and trimethoprim (dfrG). Chromosomal point mutations were also observed in gyrA, grlA, and grlB, conferring resistance to quinolones. Additionally, several efflux pump genes were identified [arlR, arlS, mgrA, norA, norB, lmrS, tet(38)], further contributing to the multidrug resistance profile of these strains.
The virulome of both strains included genes involved in adherence (e.g., sdrC, sdrE, clfA, clfB), biofilm formation (ica operon), immune evasion (capsule synthesis genes cap8A‐cap8P), and toxin production (e.g., hlgA, hlgB, hlgC, hla, hld, hlb). These virulence genes are part of the typical accessory genome of S. aureus and are associated with colonization and infection capacity (Table 2). The virulence potential of MRSA strains was assessed using a Galleria mellonella killing assay. Despite an extensive virulome, both strains exhibited lower mortality rates compared to the virulent strain AD7a587, which is associated with human clinical infections. Specifically, the tested strains caused 20% and 30% mortality within 120 h, while strain AD7a587 resulted in 80% mortality over the same period (Figure 3).
Kaplan–Meier survival plots of Galleria mellonella larvae infected with 2 × 106 UFC/larvae of S. aureus MRSA 271 and 275 strains. Plots show an average of three independent experiments with 10 larvae per group with mortality monitored daily for 120 h. The clinical strain S. aureus AD7a587 was used as a virulent control, and S. aureus ATC + C 25923 as a non‐virulent control.
The MRSA isolates from São Camilo's wild birds displayed a close genetic relationship with MSSA strains from swine in Paraíba, northeastern Brazil, and with previously reported S. aureus ST398 strains from swine, humans, and felines in the United States. The phylogenetic analysis identified between 0 and 166 single‐nucleotide polymorphism (SNP) differences among the strains; notably, the São Camilo isolates from wild birds differed by only 27 SNPs. Furthermore, fewer than 80 SNPs separated these bird isolates from the Brazilian swine‐associated MSSA strains. These findings suggest potential cross‐species transmission and underscore the genetic proximity between wild bird and livestock‐associated strains (Figure 4, Appendix 3).
Phylogenetic tree of 21 Staphylococcus aureus ST398 isolates, showing their genomic relatedness, host species, country of origin, methicillin resistance (MRSA), antimicrobial resistance profiles, and resistance genes. Colored boxes indicate the host (e.g., swine, human, bird) and the country (Brazil or USA) where each strain was isolated. Red and green squares show MRSA and MSSA status, respectively. The heatmap displays phenotypic resistance to 19 antibiotics (red = resistant; white = sensitive) and the presence of 18 resistance genes (blue = present; white = absent), allowing comparison between resistance profiles and genetic similarity among isolates.
Discussion
4
São Camilo, a 385.34‐hectare remnant of Seasonal Semideciduous Forest within the Atlantic Forest biome, holds exceptional ecological value as one of the few preserved forest fragments in western Paraná. Its high bird biodiversity, comprising over 220 species, underscores its role as a reservoir of complex ecological interactions and a potential interface for pathogen exchange between wildlife, livestock, and the environment (Rocha Ribas et al. 2023). However, like many remnants of the Atlantic Forest, São Camilo faces increasing pressure from surrounding agro‐industrial activities (Kramer et al. 2023). The proximity to intensive livestock farming and large‐scale monocultures likely contributes to environmental contamination with resistant bacteria and antimicrobial residues, creating selective pressures that may facilitate the emergence or persistence of resistant strains in local fauna. The detection of both MRSA and MSSA in wild birds from this fragment reinforces the role of avifauna as sentinels for environmental dissemination of Staphylococcus aureus.
This scenario highlights the urgent need to better understand the dynamics of antimicrobial resistance (AMR) in wild bird populations, particularly within ecologically sensitive and human‐impacted regions such as São Camilo. A more comprehensive understanding of AMR in Staphylococcus from wild birds in Brazil is hindered by the scarcity of epidemiological and molecular data. However, key studies have identified the presence of resistant Staphylococcus spp. in diverse contexts, including illegally trafficked birds (Matias et al. 2018), urban environments (Ewbank et al. 2021; Ribas et al. 2025), and even within protected conservation areas (Saraiva et al. 2021; Ribas et al. 2025). These findings, although fragmented, point to a broader and potentially silent dissemination of resistance that may be going largely undetected. Our study builds on this emerging body of work by incorporating both phenotypic and genomic data from wild birds sampled in a natural forest fragment, thereby contributing new insights into the environmental spread of antimicrobial resistance in Brazil.
In this study, S. aureus was isolated from 12.2% of sampled wild birds, a prevalence comparable to the global average of 10.3% reported by Abdullahi et al. (2021). Moreover, S. aureus was identified in 37% of the sampled bird species (20 species across different orders and families), suggesting substantial host diversity and ecological plasticity of this bacterium in wild bird populations. Although sampling was conducted over nonconsecutive days spanning multiple months, S. aureus was detected in every sampling period, suggesting that the presence of this bacterium is not restricted to a specific season. This temporal consistency strengthens the reliability of our prevalence estimates. Nevertheless, we recognize that certain ecological or microhabitat‐level factors, such as bird movement patterns or local environmental conditions, could influence detection rates and merit further investigation in future studies.
Interestingly, most birds harboring S. aureus in our study were non‐migratory species, which reinforces the hypothesis that antimicrobial‐resistant strains, or those carrying virulence factors of anthropogenic origin, were likely acquired through local or regional exposure. This finding corroborates that resident birds may act as sentinels of environmental contamination in natural and protected areas influenced by human activity (Ribas et al. 2025). Although they do not undertake long‐distance migrations, these species often move across fragmented landscapes, such as forest edges, agroecosystems, and peri‐urban zones, potentially facilitating short‐range dissemination of resistant bacteria. In this context, a variety of environmental reservoirs and transmission vectors, such as synanthropic rodents (Devanathan et al. 2024), marsupials (Santana et al. 2022), nonhuman primates (Sales et al. 2024), water sources (Tsai et al. 2020), insects (Gwenzi et al. 2021), livestock (Ramos and Cunha 2024), and human visitors (Chong et al. 2020), may contribute to the circulation of S. aureus in these ecosystems. Despite this complexity, comprehensive environmental surveillance studies remain scarce in Brazil, particularly those assessing multiple environmental matrices. This gap highlights the urgent need for integrated One Health approaches to better elucidate the ecological dynamics of S. aureus transmission in protected and natural environments.
These ecological dynamics, shaped by the limited mobility of resident bird species and the influence of surrounding anthropogenic pressures, may not only facilitate the transmission of S. aureus across environmental interfaces but also create selective conditions that favor the emergence and persistence of resistant phenotypes. In this context, among MSSA isolates, antimicrobial susceptibility testing revealed a high prevalence of inducible MLSB phenotype. Although resistance to clindamycin and erythromycin, often linked to the erm gene family, has been documented in S. aureus from wildlife (Wardyn et al. 2012; Ruiz‐Ripa et al. 2019), the elevated occurrence of this phenotype in our study is noteworthy. Interestingly, common resistance determinants such as ermA, ermC, and ermT were not detected, suggesting the involvement of alternative resistance mechanisms, less common erm variants, or even mutations in these genes that may have hindered PCR detection. In this context, further genomic investigation will be essential to elucidate the underlying resistance determinants.
β‐lactamase production mediated by the blaZ gene was identified in 41% (n = 12) of S. aureus isolates, surpassing the 28% reported in Staphylococcus spp. from trafficked birds in Rio de Janeiro (Matias et al. 2018). These enzymes, commonly detected in clinical and livestock isolates (Neelam et al. 2022; Di Gregorio et al. 2023), are increasingly reported in wildlife (Gómez et al. 2015; Martínez‐Seijas et al. 2023). Their detection in a natural forest remnant emphasizes the environmental penetration of resistance traits typically associated with anthropized ecosystems.
Another significant finding was the detection of the scn gene in all MSSA isolates exhibiting the iMLSB phenotype and penicillinase production. scn encodes the Staphylococcal Complement Inhibitor (SCIN), a component of the immune evasion cluster (IEC) characteristic of human‐adapted S. aureus (De Jong et al. 2018). Its presence in wildlife suggests possible zooanthroponotic transmission, echoing similar concerns raised by other authors (Abdullahi et al. 2021; Gómez et al. 2021).
The oropharyngeal MRSA carriage rate in birds (1%) was slightly lower than global reports (3.4%; Abdullahi et al. 2021). Notably, MRSA was identified in Manacus manacus (non‐migratory, frugivorous) and Cyanoloxia glaucocaerulea (partially migratory, omnivorous) (Wilman et al. 2014; Somenzari et al. 2018), expanding the known host range of MRSA‐ST398 in neotropical avifauna.
Genomic analyses revealed that both strains belonged to sequence type ST398, a lineage historically associated with livestock, particularly swine (Zarazaga et al. 2018; Silva et al. 2023). ST398 is recognized for its ecological versatility and capacity to colonize multiple host species across environmental interfaces, representing one of the S. aureus lineages with the greatest zoonotic potential (Graveland et al. 2011; Haag et al. 2019). The resistome of these isolates included the ermT gene (conferring cMLSB resistance) and the tetM gene (linked to Tn916), while lacking key IEC genes such as scn, sea, sep, and chp, a genomic profile commonly reported in livestock‐associated strains (Price et al. 2012).
Phylogenetic analysis revealed close relatedness to MSSA strains isolated from swine in northeastern Brazil (Santos et al. 2021), raising the hypothesis that the MRSA isolates detected in birds may have originated from livestock‐associated MSSA strains, which later acquired SCCmec elements, possibly under antimicrobial pressure. Although the genomic data, strain characteristics, and local livestock production (Machado et al. 2025) strongly suggest a link between the isolates from wild birds and livestock‐associated lineages, the directionality of this transmission remains uncertain, and alternative pathways cannot be excluded. In this context, further genomic and metagenomic studies, including environmental samples, livestock (especially swine), humans, and other wild animals, will be essential to clarify the evolutionary trajectory and transmission dynamics.
Their subsequent detection in wild birds may reflect a dynamic interface between agriculture and wildlife, underscoring the environmental dissemination of resistance. To our knowledge, this represents the first report of MRSA‐ST398 in wildlife in Brazil. Until recently, ST398 was rarely reported in the country, with sporadic detection in livestock and occasional human infections (Silva et al. 2014; Lima et al. 2017). However, its increasing presence in urban and clinical settings, including individuals with no livestock exposure (André‐Neto et al. 2017), suggests wider dissemination. In wildlife, a study involving neotropical primates also reported MSSA isolates belonging to this clonal complex, highlighting its dissemination in Atlantic Forest fragments near urbanized areas (Sales et al. 2024). Recent genomic studies have confirmed clonal complex 398 (CC398) as the dominant MSSA lineage in Brazil (Di Gregorio et al. 2023). Alarmingly, these strains have shown the capacity to acquire SCCmec and virulence genes, especially during periods of heightened antimicrobial use, such as the COVID‐19 pandemic.
Although this study focuses on a single forest fragment, it reveals broader implications for other Atlantic Forest remnants under similar anthropogenic pressure. Hundreds of forest patches in Brazil may be facing comparable microbial threats yet remain unexplored. The co‐circulation of virulent MSSA and adaptive MRSA strains with zoonotic potential in wildlife within a protected area raises significant concerns for public and environmental health. These findings underscore the urgency for expanded One Health surveillance strategies to monitor antimicrobial resistance at the human‐animal‐environment interface.
Conclusion
5
This study highlights the emergence of antimicrobial‐resistant S. aureus, including livestock‐associated MRSA‐ST398, in wild birds from a protected Atlantic Forest fragment. The detection of resistance and virulence traits in a protected area under pressure points to the silent spread of anthropogenic microbial threats across ecological boundaries, a phenomenon that may be even more pronounced in smaller, unprotected forest fragments, an all‐too‐common scenario across multiple biomes in Brazil. These findings stress the urgent need to incorporate wildlife into antimicrobial resistance surveillance frameworks, particularly in biodiversity hotspots increasingly influenced by human activity.
Author Contributions
Mateus Rocha Ribas: conceptualization, data curation; investigation; methodology; visualization; writing – original draft; writing – review and editing. Felipe Vásquez‐Ponce: data curation; formal analysis; software; investigation; writing – review and editing. Rodrigo Cardoso: data curation; formal analysis; software. Dany Mesa: data curation; formal analysis; software. Gustavo Rocha: investigation; data curation; writing – review and editing. Victor Felipe Wolleck: investigation; data curation; writing – review & editing**. Juliana Lemos Dal Pizzol:** investigation; data curation; writing – review and editing. Izadora Borgmann Frizzo de Assunção: investigation; visualization; data curation; writing – review and editing. Vinícius Pais e Oliveira: investigation; data curation. Gabriel Salvador: investigation; data curation. Amanda Tfardoski Rodrigues: investigation; data curation. Gregory Batista Melocco: data curation; formal analysis; software. Fernanda Esposito: data curation; formal analysis; software. Johana Becerra: investigation; data curation; formal analysis. Fabienne Antunes Ferreira: resources, validation. Nilton Lincopan: resources, validation. Thaís Cristine Marques Sincero: resources, supervision; validation. Jussara Kasuko Palmeiro: resources, validation; visualization; project administration; supervision; funding acquisition; writing – review and editing. Sheila Rezler Wosiacki: supervision; validation. Silvia Cristina Osaki (in memoriam): conceptualization; investigation; funding acquisition; methodology; resources, validation; visualization; project administration; supervision.
Ethics Statement
This study was authorized by the Sistema de Autorização e Informação em Biodiversidade (SISBIO) under License #75086‐3 and approved by the Comissão de Ética no Uso de Animais—UFPR—Setor Palotina (CEUA/Palotina—Protocol No. 09/2020). The captures in São Camilo State Park were authorized by the state environmental agency Instituto Água e Terra (Protocol No. 17.317.251‐6).
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdullahi, I. N. , R. Fernández‐Fernández , G. Juárez‐Fernández , et al. 2021. “Wild Animals Are Reservoirs and Sentinels of Staphylococcus aureus and MRSA Clones: A Problem With ‘One Health’ Concern.” Antibiotics (USSR) 10, no. 12: 1556. 10.3390/antibiotics 10121556.PMC 869873034943768 · doi ↗ · pubmed ↗
- 2Algammal, A. M. , H. F. Hetta , A. Elkelish , et al. 2020. “Methicillin‐Resistant Staphylococcus aureus (MRSA): One Health Perspective Approach to the Bacterium Epidemiology, Virulence Factors, Antibiotic‐Resistance, and Zoonotic Impact.” Infection and Drug Resistance 13: 3255–3265. 10.2147/IDR.S 272733.33061472 PMC 7519829 · doi ↗ · pubmed ↗
- 3André‐Neto, E. D. , R. F. A. Pereira , R. E. Snyder , et al. 2017. “Emergence of Methicillin‐Resistant Staphylococcus aureus From Clonal Complex 398 With No Livestock Association in Brazil.” Memórias do Instituto Oswaldo Cruz 112, no. 9: 647–649. 10.1590/0074-02760170040.28902291 PMC 5572451 · doi ↗ · pubmed ↗
- 4Andrews, S. 2010. Fast QC: A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 5Argimón, S. , S. David , A. Underwood , et al. 2021. “Rapid Genomic Characterization and Global Surveillance of Klebsiella Using Pathogenwatch.” Supplement, Clinical Infectious Diseases 73, no. Suppl_4: S 325–S 335. 10.1093/cid/ciab 784.34850838 PMC 8634497 · doi ↗ · pubmed ↗
- 6Bankevich, A. , S. Nurk , D. Antipov , et al. 2012. “SP Ades: A New Genome Assembly Algorithm and Its Applications to Single‐Cell Sequencing.” Journal of Computational Biology 19, no. 5: 455–477. 10.1089/cmb.2012.0021.22506599 PMC 3342519 · doi ↗ · pubmed ↗
- 7Blanco‐Peña, K. , F. Quesada‐Alvarado , D. Salas‐González , et al. 2024. “A Multidisciplinary Approach to Analyze the Antimicrobial Resistance in Natural Ecosystems.” Environmental Research 251: 118549. 10.1016/j.envres.2024.118549.38412915 · doi ↗ · pubmed ↗
- 8Bonnedahl, J. , and J. D. Järhult . 2014. “Antibiotic Resistance in Wild Birds.” Upsala Journal of Medical Sciences 119, no. 2: 113–116. 10.3109/03009734.2014.905663.24697355 PMC 4034547 · doi ↗ · pubmed ↗