Recurrent c.‐11C>T change located upstream of the normal ATG initiation codon of ANKH causes self‐limited familial infantile epilepsy
Josua Kegele, Hendrik Juenger, Harald Frantzmann, Dieter Gläser, Marc Sturm, Holger Lerche, Tobias B. Haack, Ingrid Bader

TL;DR
A specific genetic change in the ANKH gene is linked to a rare form of infantile epilepsy that resolves by age 4 and runs in families.
Contribution
This study reports the second family with autosomal dominant infantile epilepsy caused by the ANKH c.-11C>T variant and highlights its association with SeLFIE.
Findings
ANKH c.-11C>T variant causes autosomal dominant infantile epilepsy with early onset and spontaneous resolution.
The epilepsy phenotype is consistent with SeLFIE and shows low phenotypic heterogeneity across families.
ANKH-associated epilepsy should be considered in infants with familial chondrocalcinosis or joint pain history.
Abstract
Pathogenic ANKH variants are a known cause of chondrocalcinosis (Online Mendelian Inheritance in Man [OMIM] #118600) and craniometaphyseal dysplasia (OMIM #123000). Here, we describe the phenotype and genotype of autosomal dominant infantile epilepsy caused by a c.‐11C>T change upstream of the gene's normal ATG initiation codon of ANKH in a family of southern Italian descent; we correlate the phenotype with known epilepsy syndromes and provide the first evidence of recurrence of this particular ANKH variant. Phenotyping and genotyping (short‐read exome/genome sequencing) was performed on six members of a family with self‐limited familial infantile epilepsy (SeLFIE). We describe a family with six individuals who presented with infantile onset epilepsy. All affected family members experienced focal and/or bilateral tonic–clonic seizures, sometimes triggered by fever or infection, with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
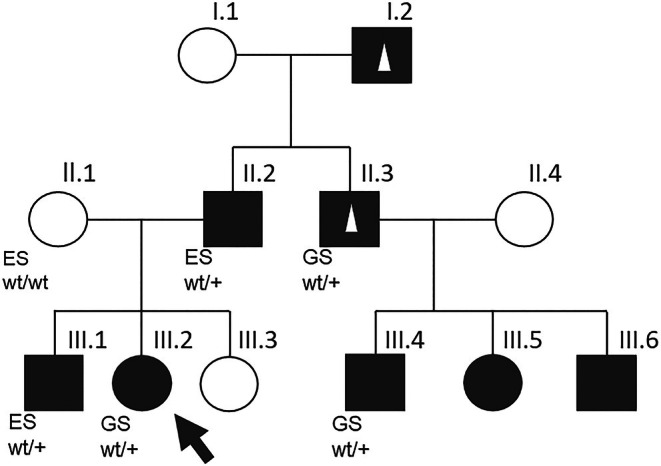 FIGURE 1
FIGURE 1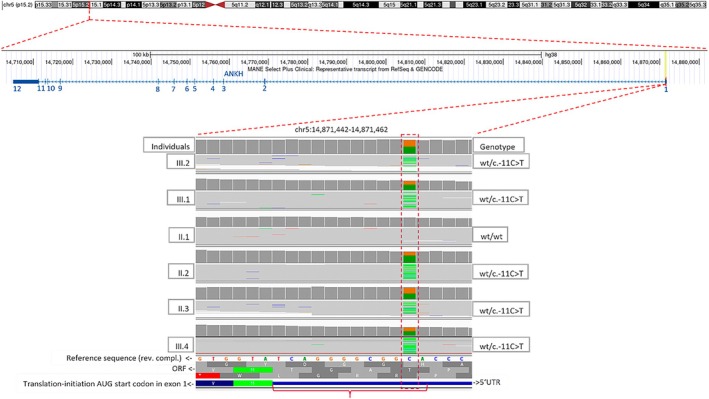 FIGURE 2
FIGURE 2| Index (III.2) | Brother (III.1) | Father (II.2) | Grandfather (I.2) | Uncle (II.3) | Cousin (III.4) | |
|---|---|---|---|---|---|---|
| ANKH (ENST00000284268.8) | c.‐11C>T | c.‐11C>T | c.‐11C>T | No genetic testing | c.‐11C>T | c.‐11C>T |
| Genomic coordinate (hg38) | chr5:14871458G>A | chr5:14871458G>A | chr5:14871458G>A | chr5:14871458G>A | chr5:14871458G>A | chr5:14871458G>A |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | No genetic testing | Heterozygous | Heterozygous |
| Gender | Female | Male | Male | Male | Male | Male |
| Age | 7 years | 8 years | 35 years | 68 years | 39 years | 12 years |
| Pregnancy, delivery | Placenta abruption; cesarean section at 32 + 1 week of gestation | Stalled birth, cesarean section at 40 + 2 week of gestation | Normal at 39th week of gestation | – | – | – |
| Length/weight/OFC at birth (centile) | 42 cm (9th–25th)/1803 g (50th−75th)/28 cm (9th−25th) | 3530 g (85th)/54 cm (35th)/33 cm (3rd) | 53 cm (91st–98th)/3910 g (91st)/35 cm (50th–75th) | |||
| Height/weight/head circumference at age of last examination (centile) | 5 years of age (18th/31st/56th) | – | – | – | – | – |
| Epilepsy type | Focal genetic epilepsy | Focal genetic epilepsy | Unclassified | Unclassified | Unclassified | Unclassified |
| Seizure type | Focal tonic nonaware seizures, also cluster of seizures | Focal to BTCS | Unclassified | Unclassified | Febrile seizure only | Unclassified |
| Age at investigation | 7 years | 8 years | 35 years | – | – | – |
| Age at onset of seizures | 10 months | 13 months | 1 year | <5 years | <1 year | – |
| Age at last seizure | 3 years (provoked) | 3 years (provoked) | 1 year | <5 years | <1 year | – |
| Seizure outcome | Seizure‐free without medication | Seizure‐free without medication | Seizure‐free without medication | Seizure‐free without medication | Seizure‐free without medication | – |
| Trigger | Infection | Fever, afebrile infection | None | None | Fever | – |
| Development | Normal milestones | Normal milestones | Normal | – | Normal | – |
| Antiseizure medications | PB, VPA | PB, LEV, BRV, good response | PRM | None | PRM | – |
| Duration of antiseizure medication treatment | 12 months (10–22 months of age) and 4 years (22 months of age until 6 years of age) | 2 years (1–3 years of age) and 3 years after encephalitis | 2 years | None | “Several years” | – |
| Comorbidities | Hemangioma, parieto‐occipital right (extracranial); rotavirus gastroenteritis at age of 3 years triggering seizures; transient ataxia | Encephalitis at age 3; postencephalitis: mild delayed motor and speech development, not fully recovered | None | Rheumatoid disease with joint pain and common use of ibuprofen, no exact diagnosis available, age at onset not known | Chronic pain in the knees, starting at the age of 30 years, taking painkillers regularly, also suffers from joint pain attacks in the knees | – |
| Neurological examination | Mild fine motor deficit and coordination deficit | Mild fine motor deficit, logopedic therapy, bad school performance | Normal | – | – | – |
| EEG | Normal | Normal | – | – | – | – |
| cMRI | Accentuation of the inner and outer liquor systems and minimal paraventricular gliosis, no epileptogenic lesion | Normal (during encephalitis T2 hyperintensities in the trigonum collaterale and the splenium, follow‐up MRI 4 years later: normal) | – | – | – | – |
- —University of Tübingen10.13039/501100002345
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Rare Diseases · RNA and protein synthesis mechanisms · Metabolism and Genetic Disorders
Key points
- Here, we report the second family with autosomal dominant self‐limited infantile onset epilepsy associated with the ANKH c.‐11C>T variant.
- Our work provides evidence for the recurrence of the ANKH variant c.‐11C>T and that this particular variant is causal for epilepsy in humans.
- The phenotype observed in affected individuals can be classified as SeLFIE.
INTRODUCTION
1
The human ANKH‐gene is located on the short arm of chromosome 5 in 5p15.2. Pathogenic variants in ANKH (Online Mendelian Inheritance in Man [OMIM] #605145) have been associated with craniometaphyseal dysplasia1 (OMIM #123000) and autosomal dominant inherited chondrocalcinosis 2 (CCAL2; OMIM #118600) in humans.2 CCAL2 is characterized by the deposition of calcium pyrophosphate crystals in the joints and manifests with episodes of acute arthritis or chronic arthropathy with age at onset typically in the third decade. The original publication, which for the first time described human ANKH variants that were associated with autosomal dominant chondrocalcinosis (CC), reported three families carrying three different ANKH variants (‐11C>T, M48T, and E490del). A UK family, in which a c.‐11C>T change created a novel ATG initiation codon in the 5′ untranslated region (UTR) of ANKH, was unique in that adult onset CC was preceded by repeated self‐limiting seizures during childhood. The N‐terminal extra four amino acids extension found in the UK family was hypothesized to produce a new activity or interaction in neural cells, a known site of ANK expression in mice and humans.2, 3 In all 13 affected members of the UK family, the c.‐11C>T variant segregated with adult onset CC preceded by febrile seizures and/or early onset and self‐limiting epilepsy. To our knowledge, no other individuals with ANKH‐associated autosomal dominant epilepsy have been reported in literature since then.
Here, we describe the second family with ANKH‐related self‐limited autosomal dominant infantile epilepsy.
MATERIALS AND METHODS
2
Written informed consent was obtained from individuals II.1, II2., III.1, III.2, II.3, and III.4 to participate in the study and to publish clinical and genetic information. The study was conducted according to local regulations and was approved by the ethics committees involved (ethics committee of the Faculty of Medicine at University Hospital Tübingen, Germany, project number 198/2010BO1). All study‐related procedures were performed in accordance with the ethical standards outlined in the Declaration of Helsinki and its later amendments.
A comprehensive personal clinical assessment was conducted on three affected individuals (III.1, III.2, and II.2). Clinical data concerning individuals I.2, II.3, and III.4 were obtained through interviews with family members II.1 and II.2.
Clinical data were collected by the investigators directly from the patient, from medical charts, or from the physician in charge. For genetic testing, EDTA‐blood samples were taken and DNA was extracted using standard procedures.
Regarding genetic testing, for patients II.1, II.2, and III.1, trio‐exome sequencing was performed from genomic DNA extracted from peripheral blood. Coding regions and adjacent intronic regions were enriched using a SureSelect XT Human All Exon kit v6 (Agilent Technologies) for subsequent sequencing as 2 × 100‐bp paired‐end reads on a NovaSeq6000 system (Illumina).
Additional genome sequencing was performed on genomic DNA extracted from peripheral blood for patients II.3, III.2, and III.4. Genomic DNA was processed with the TruSeq PCR‐Free Library Prep Kit (Illumina) and subsequently sequenced as 2 × 150‐bp paired‐end reads on a NovaSeq6000 system (Illumina).
Generated sequences were analyzed using the megSAP pipeline (https://github.com/imgag/megSAP). Clinical variant prioritization included several filtering steps including a search for rare (minor allele frequency < .1% in gnomAD and an in‐house database) variants in genes that have been associated with the patient's phenotype according to an in‐house standard operating procedure. Clinical variant prioritization was conducted independently by two trained diagnostic molecular geneticists.
RESULTS
3
Clinical data
3.1
Patient III.2
3.1.1
Delivery of III.2 was premature by emergency cesarean section due to placental abruption. Birth weight of the female was 1800 g, and neonatal intensive care/continuous positive airway pressure treatment was necessary. After 10 days, the neonate was discharged with a body weight of 2000 g. Development was normal and uneventful except for a respiratory syncytial virus infection at 3 months of age. At age 10 months, she was admitted to the hospital because of a first unprovoked seizure. Another generalized tonic–clonic seizure was observed upon admission, prompting the initiation of phenobarbital treatment (11.6 mg/kg). One year later, an attempt was made to gradually discontinue phenobarbital, but shortly thereafter, she experienced a series of seizures, comprising atonic and generalized tonic–clonic seizures, necessitating the initiation of a maintenance therapy with valproate (300 mg/day, 25 mg/kg, serum level = 54 mg/L).
At age 30 months, she experienced recurrent focal nonaware motor seizures, particularly tonic seizures of the right arm, provoked by a rotavirus infection. Cerebral magnetic resonance imaging (MRI) at 33 months of age showed accentuation of the inner and outer liquor systems and minimal paraventricular gliosis, with no epileptogenic lesion detected. Repetitive electroencephalography (EEG) yielded normal results. Antiseizure medication was stopped at the age of 5 years, and she remained seizure‐free.
At the last follow‐up, at 7 years of age, she was still seizure‐free without medication but was receiving logotherapy and ergotherapy. In terms of cognition, according to the mother, she demonstrated superior performance compared to her affected brother (Figure 1; III.1) but lagged behind her unaffected younger sister (Figure 1; III.3). She commenced schooling at 6 years 11 months of age. Neurological examination revealed discrete fine motor and coordinative deficits but was otherwise unremarkable. The EEG showed an 8/s alpha rhythm with normal background activity and no epileptiform discharges. Her growth parameters were normal at the last follow‐up at the age of 5 years (Table 1). The family history was positive for epilepsy, indicating an autosomal dominant inheritance pattern (Figure 1). The clinical information for all affected individuals in the family is summarized in Table 1.
Pedigree of the affected family carrying the variant ANKH c.‐11C>T. Squares = males; circles = females; white squares/circles = unaffected; black squares/circles = affected by infantile seizures; small white triangle = affected by joint pain/chondrocalcinosis in adulthood; black arrow = index patient (III.2). ES, exome; GS, short‐read genome sequencing performed in this patient; wt, wild‐type allele; +, allele carrying the variant ANKH c.‐11>T.
Sequence analysis, variant interpretation, databases, and cosegregation analyses
3.2
Genetic analysis revealed the heterozygous variant ANKH (ENST00000284268.8):c.[‐11C>T];[=] p.[?];[=] in all affected tested individuals, which creates an alternative start‐codon in the 5′UTR of exon 1 (see Figure 2). The variant is not present in the genomes of a control database in 151 942 alleles (gnomAD v4.1.0) but cosegregates with the disease in all five tested individuals of our family (Figures 1 and 2).
Top: Sketch of chromosome 5. Middle: Zoom‐in to 5p15.2 of the reference‐sequence (hg38) showing the genomic organization of all 12 exons of the MANE transcript (ENST00000284268.8) of ANKH as annotated in the UCSC Genome Browser. Bottom: Zoom‐in to exon 1 of ANKH and screenshot from the Integrative Genomics Viewer showing the reads at the position with the heterozygous variant ANKH c.‐11C>T (dashed rectangle) in the 5′ untranslated region (UTR) from six individuals of the family. The C>T change leads to an alternative ATG (AUG) start‐codon, elongating (bracket) the N‐terminal open reading frame (ORF) for four additional residues (Met‐Ala‐Gly‐Thr). The mother (II.1) does not carry the variant. rev. compl, reverse complementary; wt, wild‐type.
Currently, 19 different pathogenic ANKH variants are listed in the Human Gene Mutation Database (HGMD Professional 2024.4, March 12, 2025): 11 missense variants, one splice variant, five small in‐frame deletions, one small indel, and one regulatory variant (which is the c.‐11C>T change). All of the listed variants are associated with either craniometaphyseal dysplasia (OMIM #123000) or CCAL2 (OMIM #118600). This is also true for the current ClinVar entry concerning the c.‐11C>T change (variation ID: 5196). We resubmitted the variant in ClinVar (submission ID: SUB15160455) with the observed seizure phenotype (Orpha number: 306).
DISCUSSION
4
Here, we report the second family with autosomal dominant self‐limited infantile onset epilepsy associated with ANKH c.‐11C>T. The epilepsy‐related phenotype can be summarized as epilepsy with focal and/or bilateral tonic–clonic seizures with seizure onset predominantly before 2 years of age. Seizures are sometimes triggered by infection and/or fever. Patients respond well to antiseizure medication, and seizures resolve completely by 5 years of age. The phenotype observed in this family resembles that of a previously reported British family with 13 affected members,4 suggesting low phenotypic heterogeneity. In the British family, seizure onset occurred within a similar range (between 6 months and 2 years); the offset of seizure has even been described as at the age of 4 years at the latest in all affected individuals. Fever‐associated seizures were also documented. Seizure clusters have been observed in both families (the UK family and the family of Italian descent described here) as well as encephalitis with subsequent learning disability in one member of each family. Mild learning disability has been documented in three affected individuals of the British family, which is in line with the neurocognitive abnormalities diagnosed in our patients III.1 and III.2.
The phenotype of the affected members of our family shows close similarity with self‐limited familial infantile epilepsy (SeLFIE; Orpha number: 306), formerly known as benign familial infantile epilepsy (OMIM #607745); age at onset (6 months to 2 years), seizure types (focal seizures and focal to bilateral tonic–clonic seizures), occurrence of seizure clusters, good treatment response, normal EEG and MRI, and autosomal dominant inheritance of *ANKH‐*associated epilepsy are in line with SeLFIE,5 thus the affected members of our family members may be diagnosed as SeLFIE. We suggest testing patients who are negative for common disease‐causing variants in SeLFIE (SCN2A, SCN8A, KCNQ2, PRRT2) for the ANKH c.‐11C>T change, especially if there is evidence for a familial arthropathy.
All individuals who were affected by seizures in the British family also suffered from calcium pyrophosphate deposition, suggesting a high penetrance for the arthropathy with onset in adulthood. In our family, two individuals have been reported to experience chronic (I.2) or episodic (II.3) joint pain (see also Table 1). Although an exact diagnosis was not available, the reported symptoms are consistent with ANKH‐associated CCAL2.
In vitro analyses have shown that the c.‐11C>T change creates an elongated polypeptide with four additional residues added to the N‐terminus (Met‐Ala‐Gly‐Thr) compared to the wild‐type sequence.2 Functional studies demonstrated increased activity (gain of function) of the encoded protein ANKH inorganic pyrophosphate transport regulator, thus increasing intracellular pyrophosphate.2 The pathophysiological mechanism that leads to epilepsy is not well understood and has been poorly investigated; upregulation of ANK, the rodent homolog of ANKH, has been observed following seizure induction in rats, indicating a possible role in neuronal excitation.6 It is hypothesized that alterations in intra‐ and extracellular pyrophosphate levels may influence neuronal membrane excitability, contributing to a predisposition to seizures.4 Probenecid, an anion transport inhibitor, has been proposed as a potential agent for precision therapy, as it also inhibits ANKH transport activity.3 However, it remains unclear whether the altered function of the mutated ANKH directly causes epilepsy or interacts with other, yet unidentified pathways involved in epilepsy or neurodevelopment.
Further evidence for a role of ANKH in the central nervous system comes from a consanguineous family where homozygosity of the ANKH missense variant (L244S) segregated with an autosomal recessive disorder comprising mental retardation, deafness, ankylosis, and mild hypophosphatemia.7
ANKH‐associated epilepsy may be underdiagnosed for two reasons; first, the C>T change is located in the 5′UTR region of the gene, 11 nucleotides upstream of the “normal” translation initiation start‐codon, and second because of the currently missing connection of the ANKH gene entry with a seizure phenotype in relevant databases that are integrated in standard bioinformatic pipelines for variant calling and variant interpretation (e.g., databases like OMIM and ClinVar). The identification of more epilepsy patients with this variant could potentially lead to the development of therapeutic targets aiming at the prevention not only of epilepsy but also of neurocognitive disabilities and arthropathy.
The study has several limitations. The clinical history of the affected second branch (II.3 and III.4) of the family and of the grandfather (I.2) is limited and may be affected by recall bias. Finally, the proposed core phenotype discussed here is based on data from only two families. Additional reports and long‐term follow‐up of carriers of the ANKH c.‐11C>T variant are necessary to further expand the phenotypic spectrum.
CONCLUSIONS
5
Our work provides evidence for the recurrence of the ANKH variant ‐11C>T. We provide cosegregation data and genotype–phenotype correlation that confirm that this particular variant and the related pathomechanism are causal for autosomal dominant epilepsy in humans. Our study highlights the role of ANKH in epilepsy and will help to correctly diagnose infantile epilepsy more often, by shedding new light on this potentially underdiagnosed genetic form of epilepsy.
AUTHOR CONTRIBUTIONS
Josua Kegele: Conceptualization (lead); writing—original draft (lead); formal analysis (equal); writing—review and editing (equal). Hendrik Juenger, Harald Frantzmann, and Dieter Gläser: Writing—review and editing (equal). Marc Sturm: Software (lead); writing—review and editing (equal). Holger Lerche: Writing—review and editing (equal); supervision (equal). Tobias B. Haack: Writing—review and editing (equal); supervision (equal). Ingrid Bader: Conceptualization (supporting); writing—original draft (supporting); formal analysis (lead); writing—review and editing (equal).
FUNDING INFORMATION
J.K. received funding from the University of Tübingen (Fortuene‐Antrag Nr. 301100).
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nürnberg P , Thiele H , Chandler D , Höhne W , Cunningham ML , Ritter H , et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001;28(1):37–41. 10.1038/ng 0501-37 11326272 · doi ↗ · pubmed ↗
- 2Pendleton A , Johnson MD , Hughes A , Gurley KA , Ho AM , Doherty M , et al. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002;71(4):933–940.12297987 10.1086/343054 PMC 378546 · doi ↗ · pubmed ↗
- 3Ho AM , Johnson MD , Kingsley DM . Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289(5477):265–270. 10.1126/science.289.5477.265 10894769 · doi ↗ · pubmed ↗
- 4Mc Kee S , Pendleton A , Dixey J , Doherty M , Hughes A . Autosomal dominant early childhood seizures associated with chondrocalcinosis and a mutation in the ANKH gene. Epilepsia. 2004;45(10):1258–1260. 10.1111/j.0013-9580.2004.19504.x 15461680 · doi ↗ · pubmed ↗
- 5Millevert C , Weckhuysen S , Commission for the IG . ILAE genetic literacy series: self‐limited familial epilepsy syndromes with onset in neonatal age and infancy. Epileptic Disord. 2023;25(4):445–453. 10.1002/epd 2.20026 36939707 · doi ↗ · pubmed ↗
- 6Yepes M , Moore E , Brown SAN , Hanscom HN , Smith EP , Lawrence DA , et al. Progressive Ankylosis (Ank) protein is expressed by neurons and Ank Immunohistochemical reactivity is increased by limbic seizures. Lab Invest. 2003;83(7):1025–1032. 10.1097/01.LAB.0000075640.49586.E 6 12861042 · doi ↗ · pubmed ↗
- 7Morava E , Kühnisch J , Drijvers JM , Robben JH , Cremers C , van Setten P , et al. Autosomal recessive mental retardation, deafness, ankylosis, and mild hypophosphatemia associated with a novel ANKH mutation in a consanguineous family. J Clin Endocrinol Metab. 2011;96(1):E 189–E 198. 10.1210/jc.2010-1539 20943778 PMC 5393418 · doi ↗ · pubmed ↗