K-carrageenan fabricated γ-irradiated graphene oxide hydrogel: A versatile anticancer nanodrug carrier system
Samina SARWAR, Sumayya AZIZ, Asif RAZA, Tariq YASIN, Nuzhat SHAFI

TL;DR
This study develops a pH-sensitive hydrogel using graphene oxide and kappa carrageenan for controlled drug delivery, showing high drug release efficiency at physiological pH.
Contribution
A novel pH-responsive hydrogel system using gamma-irradiated graphene oxide and kappa carrageenan for efficient and controlled drug delivery is introduced.
Findings
Hydrogels showed high swelling at acidic and neutral pH, decreasing at basic pH.
Letrozole was released up to 99% in 4 hours in PBS, indicating effective drug release.
The hydrogel system is suitable for controlled injectable drug delivery at physiological pH.
Abstract
In this study, gamma-irradiated graphene oxide was incorporated into novel pH-responsive hydrogels. The biopolymer kappa carrageenan was blended with polyvinyl alcohol in a silane crosslinked biopolymer, and the effect of gamma irradiation dosage was studied. Nonhazardous tetraethoxysilane (TEOS) was used as a crosslinker. The hydrogels were characterized with chemical and thermal analysis, scanning electron microscopy, and structural analysis using a variety of analytical tools. The swelling behavior of fabricated hydrogels was assessed in various solution media. As the pH of the media increased, the swelling ratio of hydrogels decreased. All fabricated hydrogels had a high swelling ratio at acidic and neutral pH levels, with a decrease in swelling observed at basic pH. The pH-sensitive response at pH 7 make these hydrogels of potential use for controlled injectable-based drug…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
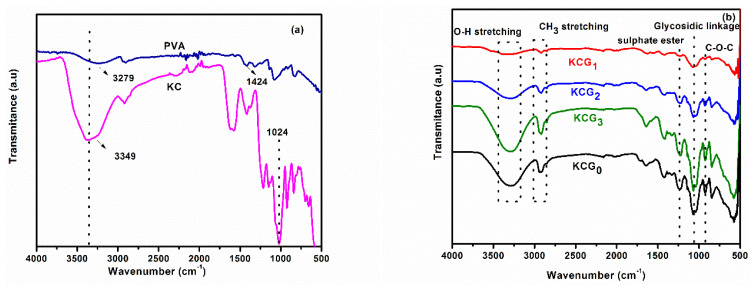 Figure 1
Figure 1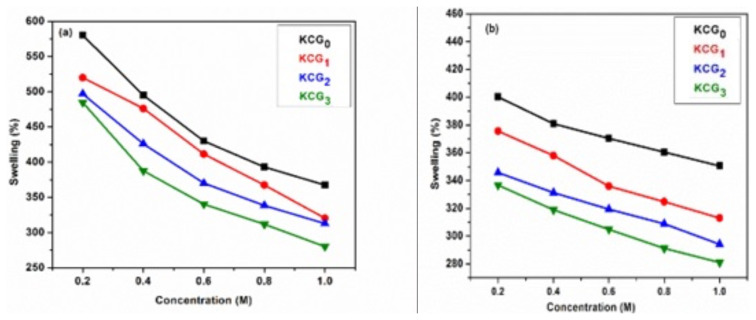 Figure 2
Figure 2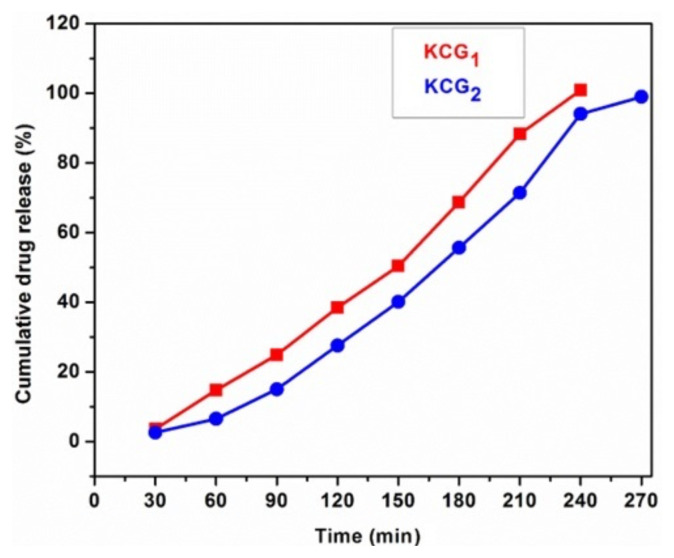 Figure 3
Figure 3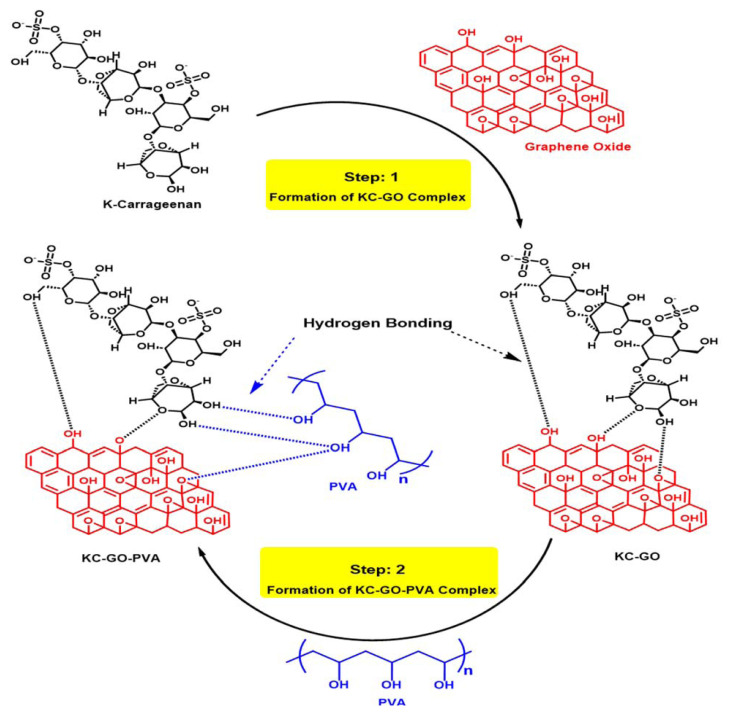 Figure 4
Figure 4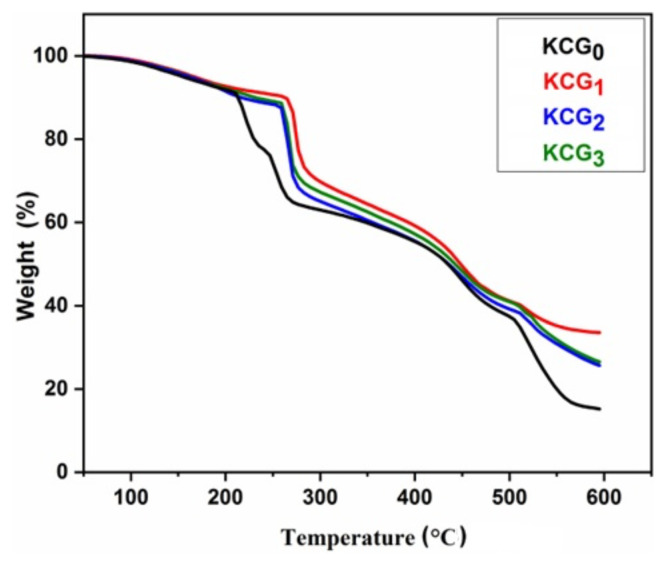 Figure 5
Figure 5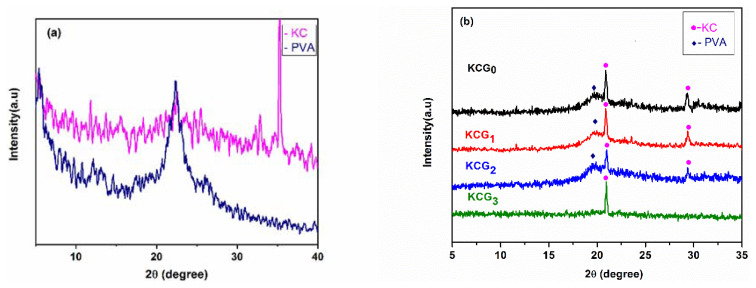 Figure 6
Figure 6 Figure 7
Figure 7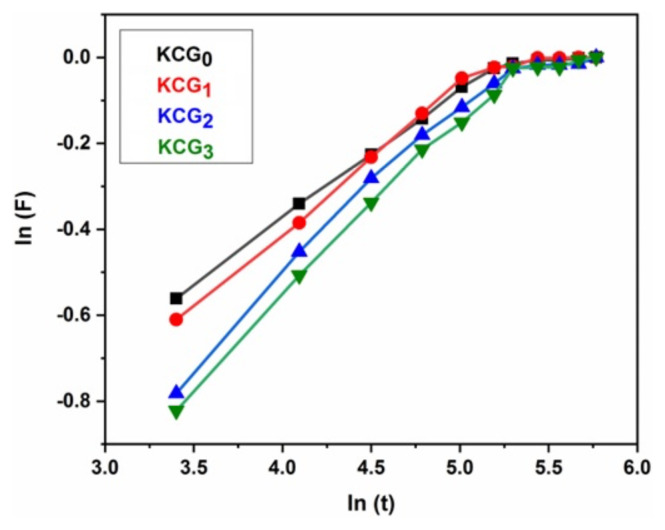 Figure 8
Figure 8 Figure 9
Figure 9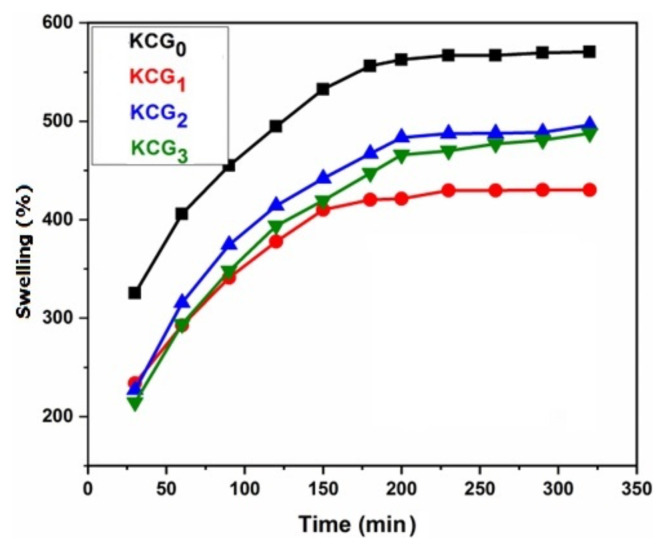 Figure 10
Figure 10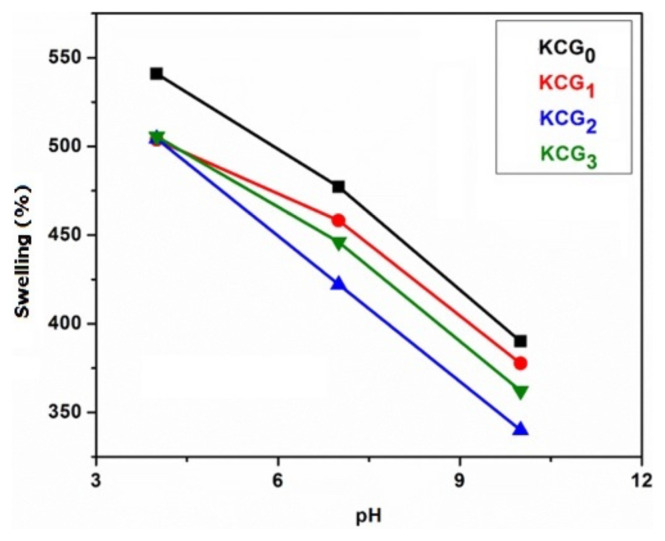 Figure 11
Figure 11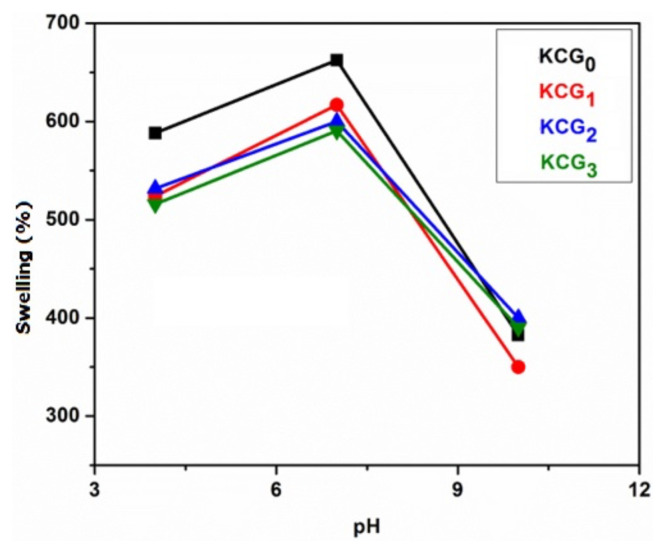 Figure 12
Figure 12Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGraphene and Nanomaterials Applications · Polymer Nanocomposite Synthesis and Irradiation · Hydrogels: synthesis, properties, applications
Introduction
The use of regulated drug-delivery systems (DDSs) has increased dramatically in the last couple of decades for better regulation of drug-release kinetics in the human body. Controlling the release of drugs helps to regulate the amount of medication in the body, shield the drug from physiological conditions that would otherwise cause it to break down and be eliminated, deliver the medication precisely with the least amount of exposure, and improve patient compliance [1]. Site-specific medicine is important for treating cancer, wound dressing, and in gene therapy. Such a system is possible with biodegradable and synthetic polymers. Nevertheless, these delivery systems can be expensive, toxic, and have side effects. Thus, there is a need for such a system to be safe and cost effective, with low side effects [2]. Smart DDSs (SDDSs) can simultaneously block signal pathways, respond to stimuli, transfer drugs, and stop releasing drugs [3]. SDDSs work on the principle of transferring a required amount of medicine at a predictable time to the affected site. The controlled signals of SDDSs are not only constrained to internal conditions (e.g., pH, the concentrations of particular biomolecules, catalytic function, and redox reactions [4]) but also influenced by external factors like the light of several wavelengths, magnetic behaviors, ultrasound, and electric fields [5,6]. The basis of SDDSs can be carrier hydrogels, polymers, nanoparticles, liposomes, or nanosheets, and the smart carriers are micelles [7–9]. Hydrogels are a network of 3-dimensional chains consisting primarily of homo- or copolymers with hydrophilic characteristics. They swell without being soluble and behave as colloids in which the dispersion phase is water. The physical or chemical crosslinks give the hydrogel a specific shape and insolubility. They are highly permeable with a water content of 99%. This, combined with their soft structure and absorbance, means hydrogels can be considered a natural living tissue [10–12]. Hydrogels have various benefits as DDSs. They have good swelling properties, biocompatibility, and less toxicity depending on their composition and process handling [13]. Hydrogels are a diverse and versatile class of material that are not only of use in drug delivery but also in fabricating eye lenses, wound dressing, and in tissue manufacturing [14].
Hydrogels can be made with natural or synthetic polymers. Natural hydrogels made of collagen, fibrin, and gelatin are biocompatible, biodegradable, and biologically distinguishable moieties that help the performance of different organs. However, such polymers are mechanically weak and may have microbes or induced inflammatory immune reactions [15,16]. Various synthetic hydrogels polymers like polyvinyl alcohol (PVA), polyacrylic acid (PAA), and polyethylene oxide (PEO) are altered to produce versatile functions and degradability but lack biological characteristics of natural polymers.
Recently, the smart behavior of hydrogels combined with the flexible and tunable characteristics of materials has further increased their scope in biomaterials research. Polymer hydrogel scaffolds are effective materials for tissue engineering in the repair and regeneration of different organs and tissues. Hydrogels have been widely applied in various disciplines in biomedicine for more than 5 decades [17,18]. The incorporation of biopolymers in hydrogel fabrication facilitates the development of biodegradable, biocompatible, and nonimmunogenic drug carriers [19].
More recently, there has been more interest in fabricating hydrogels as novel DDSs. Hydrogels have the capacity for site-specific and regulated delivery of curative mediators [20]. Hydrogels have been developed as medicine carriers and discharge-rate controllers [21–25]. Biomedically synthesized hydrogels deliver drugs accurately and can release drugs in response to external stimuli. Smart polymer hydrogels adjust their position and structure according to external stimuli and have significant potential for scientific investigation and several advanced technical uses.
The natural polymer kappa carrageenan (KC) is made up of 3,6-anhydro-D-galactose monomers and D-galactose-4-sulfate in a linear sequence. There are 3 subtypes of carrageenan, but only the iota and kappa subtypes are frequently utilized for gel formation. However, the fiber structure of carrageenan has low mechanical strength [26]. Synthetic polymers can be combined with biopolymers to enhance various properties [27]. PVA is a synthetic polymer with strong mechanical properties. Hydrogels made from biopolymers blended with PVA are reliable in their physical characteristics and biocompatibility [28,29]. PVA has been used extensively by researchers for several applications in biomedicines, sorption, water treatment, and agriculture [30,31]. Despite their remarkable properties, hydrogels face limitations in drug delivery due to low loading capacity (especially for hydrophobic drugs), poor mechanical strength, low homogeneity, and inadequate stimuli response. To overcome these challenges, 2-dimensional (2D) graphene has been integrated into 3-dimensional (3D) hydrogels, enhancing their performance. This 2D–3D synergy has enabled the development of advanced graphene-based hydrogels (GBH), where graphene acts as a gelator for self-assembly or a filler for blending with various molecules, creating multifunctional GBH with improved properties.
Since the discovery of graphene in 2004, the monolayer of carbon atoms in tight arrangement in a honeycomb-like structure has drawn attention. The 2D structure of graphene has unique mechanical, thermal, physical, and electronic properties, as well as particular magnetization and a large surface area [32–35]. Graphene structures have limited chemical reactivity due to the absence of functional groups. Recently, the surface of graphene has been enhanced with oxygen, producing a derivative that is hydrophilic and contains functional groups such as hydroxyl, carbonyl, carboxyl, and epoxy [36,37]. Ionizable carboxyl groups, present in graphene oxide (GO), may exchange with metal ions and help GO disperse in some polar liquids. This improves the interfacial interaction between GO and polar molecules, which in turn increases the mechanical characteristics of polar molecules [38]. Gamma (γ) irradiation offers a safer, faster, and more efficient method for synthesizing high-purity graphene composites under ambient conditions. γ-Irradiated, graphene-based nanomaterials have applications in catalysis, energy, sensing, and biomedical fields. Carbon materials like carbon nanotubes, carbon nanofibers, and graphene enhance the photocatalytic activity of materials. Graphene has strong antibacterial effects due to its sharp edges and high surface area that allow it to penetrate bacterial membranes, causing structural damage and leakage of cellular contents. The nanosheet structure of graphene can also disrupt the integrity of cell walls, leading to bacterial cell death. Additionally, the ability of graphene to interact with microbial DNA and proteins further contributes to its antibacterial activity. The side effects of conventional cancer treatments highlight the need for less harmful alternatives, leading to the exploration of nanomaterial-based photothermal therapy (PTT), photodynamic therapy (PDT), and bioimaging. Biocompatible GO is increasingly used in hydrogels to immobilize therapeutic agents and enhance mechanical and thermal properties. GO offers therapeutic advantages, including the ability to carry bioactive molecules and improve drug-loading efficiency [39].
Letrozole (LTZ) was chosen as a model drug in this study. It is a form of aromatase inhibitor medication that is used in the treatment of breast cancer. GO-incorporated hydrogels enable controlled and sustained drug release at the tumor site, significantly improving the therapeutic efficacy of LTZ, a poorly water-soluble drug. Postsurgical hydrogel implants are a viable alternative to traditional chemotherapy because they target leftover tumor cells, reduce recurrence risks, and minimize systemic toxicity. Future research should focus on their long-term stability, in vivo efficacy, and potential translation into therapeutic applications. Injectable GO-incorporated hydrogels are a promising drug delivery platform because of their simplicity of administration, extended drug release, and enhanced bioavailability. In the current study, tetraethoxysilane (TEOS) was used as a crosslinker to create novel injectable hydrogel blends of biopolymers (KC) and synthetic polymers (PVA) with varying doses of irradiated GO. No such combination (KC, γGO, and PVA) has been reported as a DDS before. Moreover, previous DDSs were administrated orally while this study presented drug delivery through an injectable route. LTZ was loaded into the novel hydrogels to show the controlled release in PBS. The fabricated LTZ-loaded hydrogel sample released the drug in PBS in a controlled manner.
Materials and methods
2.1. Chemicals and material
KC was acquired from Quest International (Metro Manila, Philippines). Graphite powder was purchased from Merck (Frankfurt, Germany). PVA, potassium persulfate (KPS), vinyltriethoxysilane (VTES), hydrochloric acid (HCl), sulfuric acid (H_2_SO_4_), hydrogen peroxide (H_2_O_2_), potassium permanganate (KMnO_4_), calcium chloride (CaCl_2_.2H_2_O), sodium chloride (NaCl), and TEOS were acquired from Sigma-Aldrich (Darmstadt, Germany) and used as directed. Deionized water (DIW) was used to prepare all solutions.
2.2. Synthesis of GO
GO was extracted from graphite powder using a modified Marcano method [40]. The oxidation of graphite flakes was accomplished by mixing 1 g of graphite, 4 mL of orthophosphoric acid, 40 mL of sulfuric acid, and 2 g of potassium permanganate while continually stirring the mixture with a magnetic stirrer. In order to control the exothermic reaction during the addition of potassium permanganate, the whole procedure was performed in an ice bath at 4 °C. The oxidation reaction was stopped by the addition of 10 mL of hydrogen peroxide. Oxidized graphite particles (GO) were collected and rinsed with 1 M HCl and DIW. The GO particles were then incubated in a desiccator after being dried under a vacuum for 48 h.
2.3. Irradiation of GO
The prepared GO powder was irradiated at different irradiation doses (0 kGy, 100 kGy, and 125 kGy) using a cobalt-60 irradiator (24 kCi) at a dosage rate of 5.0 kGy/h at the Nuclear Institute of Food and Agriculture (NIFA), Peshawar, Pakistan.
2.4. Synthesis of the hydrogel blend
A solution casting approach was used to develop GO-loaded hydrogel blends. A measure of 0.4 g of KC powder was dissolved in 40 mL of distilled water with constant stirring at 40 °C. A measure of 0.6 g of PVA was dissolved separately in DIW at 80 °C. Irradiated GO powder (5% w/v) was dispersed in 10 mL of DIW in separate glass beakers and sonicated for 2 h. The sonicated GO solution was mixed with the KC solution at 60 °C followed by the addition of the PVA solution under continuous stirring. Then, a 4% TEOS solution was added in a dropwise manor into the KC/PVA/GO solution and further stirred for 4 h at 60 °C. The prepared solution was then transferred to a plastic container and the solution was dried room at temperature. Dried KC/PVA/GO hydrogel films were peeled off and stored in a desiccator. Formulation and identification codes of synthesized hydrogels are represented in Table 1.
2.5. Synthesis of the drug-loaded hydrogel blend
LTZ (30 mg) was mixed in 10 mL of methanol and added in a dropwise manor to the KC/PVA/GO solution synthesized in the previous step. After continuous stirring for 1 h, the TEOS crosslinker was gradually added at a constant temperature of 60 °C. The solution was poured into a plastic container and left to air dry.
2.6. Characterization of the hydrogel
2.6.1. Fourier transform infrared (FT-IR) spectroscopy
Fourier transform infrared (FT-IR) spectrometry of all the hydrogel samples was carried out using a Nicolet 6700 FT-IR spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) in the range of 4000–500 cm^−1^.
2.6.2. Thermogravimetric analysis
Thermal analysis measurements were carried out using a SDTA 85 thermogravimetric analyzer (Mettler Toledo, Columbus, OH, USA) in an inert environment between 50 and 800 °C and heating rate of 20 °C/min.
2.6.3. Scanning electron microscopy
Scanning electron microscopy (SEM) was used to examine the morphology of hydrogel samples using a MIRA-3 instrument (Tescan, Warrendale, PA, USA). Before scanning, the samples were freeze dried and gold coated to make their surfaces conductive before scanning.
2.6.4. X-ray diffraction analysis
The crystalline structures of hydrogel samples were examined by X-Ray diffraction (XRD) using a D8 Discover diffractometer (Bruker, Billerica, MA, USA). A scan rate of 1.1/min was used to acquire the diffractograms for 2θ from 5° to 35°.
2.7. Adsorption studies
2.7.1. Swelling behavior
The swelling behavior of developed hydrogels was investigated. Dried hydrogels (100 mg) were immersed in 30 mL DIW. Hydrogel films were weighed after regular interval. The swelling ratio of the hydrogel was calculated by the formula in Equation 1.
Where W_f_ denotes the swelled weight at time t and W_i_ denotes the initial dry weight of samples.
2.8. Swelling behavior in buffer, nonbuffer, and salt solutions
Buffer and nonbuffer solutions are used to investigate the pH sensitivity of hydrogels. Buffer solutions of pH 4, 7, and 10 were obtained from Sigma-Aldrich. Nonbuffer solutions of pH 4, 7, and 10 were prepared by diluting stock solutions of NaOH (pH 14.0) and HCl (pH 1.0) with DIW. A digital pH meter was used to determine the pH of solutions. Dried hydrogel samples were placed in 30 mL of sodium chloride and calcium chloride (0.2 M to 1.0 M) solutions and their swelling behavior was assessed.
2.9. In vitro drug release
The in vitro drug release study of the LTZ drug-loaded hydrogels was carried out in phosphate buffer saline (PBS). Drug-loaded hydrogels were initially introduced into a container containing 100 mL of PBS at a temperature of 37 °C. Aliquots of 3 mL were periodically taken out of the solution to monitor LTZ release. To maintain a steady liquid level, 3mL of fresh PBS was redeposited.
The release of LTZ was measured using UV–vis spectrophotometry at 246 nm with a Perkin Elmer Lambda 40 UV–vis spectrophotometer (Waltham, MA, USA) with PBS serving as the reference. LTZ solutions containing 5 to 30 mg/mL of solvent were used to calculate the calibration curve. Using a calibration curve, the quantity of LTZ in the solution was determined to calculate the drug release ratio.
Result and discussion
3.1. FT-IR spectroscopy
FT-IR was used for chemical analysis of the samples. The obtained spectra of pure PVA, KC, KCG_0_, KCG_1_, KCG_2_, and KCG_3_ are shown in Figure 1. Observed characteristic peaks of hydrogels are summarized in Table 2. The characteristic peaks in the 3400–3000 cm^−1^ range correspond to O–H stretching vibrations. The peak at 1210–1260 cm^−1^ represents S=O stretching of sulfate groups. Peaks in the 1010–1080 cm^−1^ range are attributed to C–O–C stretching of glycosidic linkages. The characteristic peak of 3,6-anhydro-D-galactose (C–O–C stretching) is observed at 910–940 cm^−1^, while the peak at 850–840 cm^−1^ corresponds to D-galactose-4-sulfate [41]. Apart from characteristic peaks from KC, peaks characteristic of PVA were also observed in hydrogel blends at 2926 cm^−1^ in all samples of developed hydrogels (KCG_0_, KCG_1_, KCG_2_, and KCG_3_) that can be attributed to C–H alkyl stretching [42].
Comparison of FT-IR spectra of KCG_0_, KCG_1_, KCG_2_, and KCG_3_ in Figure 1b shows that all the characteristic peaks of KC, PVA, and GO are present. The O–H stretching band region (3400–3200 cm^−1^) corresponds to the hydroxyl (O–H) stretching vibrations present in both KC and PVA. In the hydrogel blend, hydrogen bonding interactions between their hydroxyl groups can lead to a broadening of this band, indicating the formation of intermolecular hydrogen bonds between the two polymers. This effect is more pronounced compared to native KC, as observed in KCG_0_.
The intensity of the peaks increases with the irradiation dose of GO. This is attributed to the presences of polar functional groups. Hydrogen bonded −OH stretching in KCG_0_, KCG_1_, KCG_2_, and KCG_3_ samples appear at 3284, 3334, 3304, and 3294 cm^−1^, respectively. Notably, the intensity of the −OH group peaks is greater in hydrogels with higher irradiation because of the abundance of polar functional groups introduced by GO [43]. The sharp bands at 1647, 1640, 1640, and 1637 cm–1 correspond to C=O stretching in KCG0, KCG1, KCG2, and KCG3 samples, respectively. The sharp band at 1647, 1640, 1640, and 1637 cm^−1^ correspond to C=O stretching. The peak intensity also increases with the irradiation dose [44]. After the incorporation of the irradiated GO into the KC/PVA composite, the stretching peak of −OH shifted to a lower wavenumber. This occurs because the irradiated GO disrupts polymer molecules via disrupting hydrogen bonding between them, and allowing the formation of new hydrogen bonds within the matrix and GO sheets. A significant increase in −OH stretching and C–O stretching is observed with higher irradiation doses [45]. The vibration in the KC biopolymer C–O–C backbone was indicated by a sharp peak at 1027 cm^−1^ and peak shifting was observed due to crosslinking of PVA and irradiated GO at 1068, 1061, 1061, and 1058 cm^−1^ in KCG0, KCG1, KCG2, and KCG3 samples, respectively. As the irradiation dose increased, the peaks shifted to a lower wavenumber. Furthermore, the peaks at 1332 and 1424 cm^−1^ confirm the existence of stretching vibrations in functional groups that contain oxygen (e.g., carboxyl, epoxy, and ester groups). The presence of all the characteristic peaks of KC, PVA, and GO in FT-IR spectra and the shifting of peaks due to the formation of hydrogen bonding with in polymer matrix confirms the KC/PVA/GO hydrogel combination was successfully synthesized [46].
3.2. Thermogravimetric analysis
Thermal analysis was performed to assess the thermal stability, applicability, and durability of the polymeric hydrogels. Figure 2 shows TGA thermograms of KCG_0_, KCG_1_, KCG_2_, and KCG_3_ samples. The KC/PVA/GO hydrogel has 4 thermal degradation stages at temperatures between 50–100 °C, 200–300 °C, 380–500 °C, and above 500 °C. The first degradation stage was associated with water evaporation with approximately 3.3% weight loss in all samples. The second degradation stage was associated with the disintegration of carrageenan sulfonate groups and PVA thermal degradation [47]. KCG_0_, KCG_1_, KCG_2_, and KCG_3_ samples lost 18%, 15%, 15%, and 16% weight, respectively, at 271.5 °C. This shows that hydrogel thermal stability improved due to the addition of irradiated GO. Similar results were reported by Mahdavinia et. al. [48]. The third degradation stage was associated with the degradation of the polymer backbone. Above 500 °C, KCG_0_, KCG_1_, KCG_2_, and KCG_3_ samples lost 18%, 14%, 12%, and 13%, respectively due to the breakdown of oxygen functional groups. This shows that the stability of hydrogels increased with the addition of irradiated GO up to 100 kGy. However, a further increase in irradiation dose up to 125 kGy decreases the thermal stability of the hydrogel, possibly due to the formation of defects in GO.
Table 3 shows temperatures at 20%, 50%, and 70% mass losses along with residual mass loss of each hydrogel. Analysis of degradation temperatures and residual mass shows that KCG_2_ is more thermally stable.
3.3. X-ray diffraction analysis
XRD is used to determine the structural characteristics, crystalline orientation, and crystalline phases in polymers. The XRD diffractogram of pure PVA, pure KC, and hydrogel samples are shown in Figure 3. Distinctive peaks corresponding to PVA are shown at 2θ = 19.9° in Figure 3a. In the diffractogram for KC, there is a weak broad peak near 2θ ≈ 20° in Figure 3a, indicating its amorphous nature [49]. The XRD pattern of GO shows an intense and sharp peak centered at 10.24° [50].
Figure 3b shows the XRD results of KCG_0_, KCG_1_, KCG_2_, and KCG_3_ hydrogel samples. Sharp diffraction peaks are observed at 2θ = 20.6° and 28° in all samples. A decrease in intensity isobserved with the increase in irradiation dose due to increased intermolecular interactions between the KC, PVA, and GO molecules. Furthermore, when GO was mixed with the PVA/KC solution, the hydrogel only displays the KC/PVA diffraction peaks; the distinctive GO peak is not present. This might be the result of the relatively low concentration of GO in the PVA/KC solution and the fact that the diffraction of GO is much weaker than that of KC/PVA [51].
The positions of the diffraction peaks shift slightly after the addition of GO to the hydrogel. Furthermore, the intensities of these peaks decreased considerably in contrast to the nonirradiated composite sample. The peak intensity decreased gradually as the irradiation dose increased. Increased molecular interaction due to the addition of irradiated GO causes a significant decrease in the degree of crystallinity of the hydrogel blends (KCG_1_, KCG_2_, and KCG_3_).
3.4. Scanning electron microscopy
SEM was used to examine the morphology of freeze-dried hydrogels. Representative SEM micrographs of hydrogels without GO (KCG_0_), with nonirradiated GO (KCG_1_), and GO irradiated to 100 kGy (KCG_2_) are shown in Figure 4. KCG_0_ had a smooth surface (Figure 4a), whereas hydrogels with GO were coarse with the appearance of wrinkles (Figure 4b and 4c). Additionally, the surface morphology of the composite hydrogels showed large holes that aided in the diffusion of molecules in the adsorption tests. The smaller, denser pores and wrinkles provide a larger specific surface that enhanced the absorption ability of the hydrogel. KCG_3_ did not show satisfactory results in swelling behavior, so SEM images are excluded. With the increase in surface roughness, hydrogel swelling increased due to increased water uptake, reduced water diffusion barriers, and increased surface area availability for water interaction.
3.5. Mechanism of hydrogel synthesis and drug release
Scheme illustrates how silane crosslinkers, PVA, and KC have both chemical and physical electrostatic attractions that contribute to the stable structure of the hydrogel. Additionally, these interactions make the hydrogel suitable for injection, highlighting its potential as an optimal carrier for drug delivery applications.
3.6. Swelling tests in various swelling media
3.6.1. Swelling kinetics
The solvent distribution from the extracellular matrix to the hydrogel determines how much swelling will occur. Equation 2 was used to validate the swelling data [52].
Where k is the rate of swelling constant and F stands for fractional swelling that can be described as in Equation 3:
The swelling at equilibrium time is denoted by W_eq_ and the swelling at time t by W_t_. The swelling properties of hydrogels in distilled water were used to calculate the values of k and n, where the solvent transport mechanism inside the hydrogels is defined by the value of n. When the value of n is less than or equal to 0.5, it indicates Fickian transport and a value between 0.5 and 1 indicates non-Fickian transport. Using the swelling values of hydrogels in water, a graph was plotted between ln t and ln F as illustrated in Figure 5. Table 4 presents the calculated values for the diffusion parameters, showing that the hydrogels have Fickian diffusion mechanisms.
3.6.2. Swelling study of hydrogels
The ability to swell is a characteristic feature of hydrogels. Hydrogels are polymeric networks with the capacity to retain large amounts of water in various percentages depending on their chemical composition. Both chemical composition and swelling media play significant roles in the swelling behavior of hydrogels. The degree to which ionic polymers swell depends on various structural parameters, includes hydrophilicity, crosslinked density, charge, concentration, and degree of ionization. Other factors that affect the degree of swelling include the valency, pH, ionic strength, and counter ions in the swelling medium [53]. A preliminary investigation into the swelling behavior of hydrogels was conducted and their dynamic swelling behaviors in distilled water are illustrated in Figure 6. Initially, water uptake in hydrogels rapidly increased and subsequently plateaued as equilibrium was reached. This trend aligns with typical hydrogel swelling behavior, where an initial swift absorption phase is followed by a slower approach to equilibrium. The swelling behavior of the hydrogel in saline solutions differed noticeably from that seen in deionized water and was influenced by the kind of salts utilized. In order to assess the impact of pH on hydrogel swelling, samples were studied at pH 4, 7, and 10.
3.6.3. Swelling ratio of hydrogel in distilled water
Hydrogels use diffusion to effectively absorb water. The attraction between the polymer chains and the surrounding media leads to diffusion. Figure 7 shows the swelling behavior of KC/PVA/GO hydrogels in distilled water. The swelling behavior of the hydrogel films vary widely. The stability time was 5 h for all hydrogel samples. The gel samples were placed in distilled water at room temperature and immersed until the gel reached its equilibrium condition of swelling. The hydrogel was removed from the medium, the extra solvent was quickly blotted off the surface with absorbent paper, and the sample was weighed and the average of 3 readings were reported. Figure 7 clearly shows the rate of swelling and water absorption are influenced by the composition of the hydrogels. All hydrogels containing GO had a decreased swelling capacity than hydrogels without GO added. The inclusion of GO nanoparticles is responsible for the decrease in water absorbency. Due to the presence of numerous functional groups in GO, the interaction between nanoparticles and polymeric chains results in increased crosslinking density, thereby decreasing swelling capacity [54,55].
Swelling of the hydrogel blends can be attributed to deprotonation of sulfate groups (−OSO_3_H) of carrageenan at pH 7, resulting in the creation of sulfate ions (−OSO_3_). Due to electrostatic repulsion caused by negatively charged sulfate ions repelling the sulfate ions on another chain, polymeric chains expand and lead to an increase in swelling [56]. The presence of the hydrophilic (−OH) group in PVA also had an impact on the swelling behavior [57].
3.6.4. Effect of irradiation dose of GO
The effect of gamma irradiation on hydrogels was calculated by its swelling behavior. The swelling curves of hydrogels containing GO samples irradiated at different doses (0, 100, and 125 kGy) are shown in Figure 6. At first, swelling increased with the addition of GO irradiated to 100 kGy. This is because of more hydrophilic groups in GO by irradiation. As more hydrophilic hydroxyl and carboxyl functional groups are available, they are more likely to interact with water molecules. However, samples containing GO irradiated at 125 kGy showed a decrease in swelling. This may be caused by the aggregation of GO and an increase in crosslinking density. This produces hydrogels with a more compact structure due to the ability of irradiated GO to trigger crosslinking reactions [58].
3.6.5. Swelling in buffer solution
The response of prepared hydrogels to buffers with pH values of 4, 7, and 10 was examined. Figure 8 shows the effect of buffer solution pH on swelling behavior of hydrogels. At acidic pH, all samples showed the maximum swelling, moderate swelling was seen at pH 7, and decreased swelling was noted at basic pH. The protonation of the OH group of PVA and −SO_3_ of the KC caused the swelling in acidic media. Higher concentrations of charged units and ionic groups in an acidic medium cause charge repulsion that raises the swelling ratio when more solvent enters by osmosis. All hydrogels, on the other hand, consistently declined in swelling as the pH change from acidic to basic, with minimal swelling in basic conditions because of a decrease in the ionization of charged units. This reduction results in the contraction of polymer chains, thereby decreasing the swelling capacity. Maximum swelling was observed by KCG_0_ among all the samples.
3.6.6. Swelling in nonbuffer
The swelling response of the hydrogels in nonbuffer solutions at pH values of 4, 7, and 10 is shown in Figure 9. Swelling of hydrogels increased with pH 4 to pH 7 due to ionization of the sulphonic acid groups, i.e. OSO_3_H that imparts negative charge to the KC back bone. Free ion concentration within the gel phase increases due to ionization. As a result, it causes an increase in swelling. All hydrogel blends showed a consistent decline in swelling from an acidic to a basic pH, due to the reduced ionization of charged units, causing polymer chain constriction and a subsequent reduction in swelling across all hydrogels [59].
3.6.7. Swelling in electrolytes
The effect of NaCl and CaCl_2_ concentration (0.2, 0.4, 0.6, 0.8, and 1 M) on adsorption was also studied and results are presented in Figure 10. NaCl is a monovalent Na^+^ salt and CaCl_2_ is a divalent Ca^+2^ salt. Figure 10a shows the effect of NaCl concentrations on swelling of hydrogels. It is evident that swelling percentage decreases with the increase in NaCl concentration from 0.2 to 1 M. The adsorption capacity of hydrogels was slightly reduced with the addition of sodium chloride compared to distilled water. As the polarity of the solution increased, hydrogel swelling capacity decreased. The neutralization of anionic sulfate (−OSO_3_) groups causes the reduction in swelling of hydrogels [60,61].
The adsorption capability of hydrogels in the CaCl_2_ solutions was noticeably lower than in the NaCl solution, as shown in the Figure 10b. This is caused by Ca ion interaction with hydrogel anionic centers. The screening effect of excessive charge present in the electrolyte solution causes a decrease in swelling ratio. These compounds can cause hydrogels to contract and, as a result, have a smaller surface area. Adsorption capability of the hydrogels decreases as surface area reduced. An increase in the molar concentration of the electrolytes also causes a decrease in the osmotic pressure between the electrolytes and the hydrogel matrix, which prevents water from entering into the hydrogel matrix [62].
3.7. Controlled release examination of LTZ
In vitro studies were carried out to study the drug release activity due to the fact that the hydrogels swell more in acidic pH, thus limiting their use for oral drug delivery. The drug release behavior of the hydrogels is closely associated with their swelling properties and a crucial aspect of hydrogel structural design. As the hydrogel swells, its mesh size expands, creating diffusion pathways that facilitate drug release. LTZ was selected as the model drug, and loaded on KCG_1_ and KCG_2_ hydrogels. KCG_3_ was not used for drug loading due low swelling. The release mechanism of these hydrogels was then examined in PBS with respect to time (t) at 37 °C. (Figure 11). The hydrogels KCG_1_ and KCG_2_ both showed sustained release of LTZ over a period of 5 h. Higher irradiation of GO (KCG_2_) caused slower drug release. In PBS, LTZ was released slowly; 99% of the whole dose was released in 4.5 h compared to 100% in 4 h by KCG_1_. Slow release may be attributed to the addition of GO, as GO has a large surface area with abundant functional groups (e.g., carboxyl, hydroxyl, and epoxy) that enable strong hydrogen bonds and π–π interactions with drug molecules. These interactions resulted in higher drug retention within the hydrogel matrix, decreasing its release. LTZ, being a poorly water-soluble drug, may have strong hydrophobic interactions with GO, further reducing its release in an aqueous environment like PBS. Therefore, the higher controlled release behavior of KCG_2_ highlight its potential for intravenous medications.
Conclusion
KC/PVA hydrogels combined with various doses of irradiated GO were developed and their chemical, morphological, thermal, swelling, and drug delivery properties were characterized. The hydrogel showed increased thermal stability, swelling behavior, and injection ability properties as irradiation dose increased. Water-retention capacity increased when GO was incorporated. KCG_2_ (100 kGy) showed maximum swelling and thermal stability. This increase in swelling facilitated a more efficient drug diffusion process by expanding the hydrogel network. The drug release from the hydrogels was primarily governed by a swelling-controlled diffusion mechanism. The presence of GO contributed to a more sustained and controlled release profile, reducing the initial burst release commonly seen in traditional hydrogels. The Fickian diffusion regulation mechanism was also studied in all hydrogels and the value of diffusional exponent n was found to vary from 0.243 in KCG_0_ to 0.068 in KCG_3_. The pH swelling response of hydrogels indicated high swelling ratio in acidic conditions and low swelling ratio in basic conditions. Maximum swelling was observed at around neutral pH, making these complexes particularly appropriate candidate for injectable DDSs. In drug loading and release investigations, KCG_1_ and KCG_2_ hydrogels were tested. LTZ was released in a sustained manner for a period of 4 h. This research highlights the significance of GO-based hydrogels for biomedical applications including drug delivery, tissue engineering, and wound healing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adepu S Ramakrishna S Controlled drug delivery systems: current status and future directions Molecules 2021 26 19 5905 10.3390/molecules 26195905 34641447 PMC 8512302 · doi ↗ · pubmed ↗
- 2Sung YK Kim SW Recent advances in polymeric drug delivery systems Biomaterials Research 2020 24 1 12 10.34133/bmr.0221 32537239 PMC 7285724 · doi ↗ · pubmed ↗
- 3Rana A Adhikary M Singh PK Das BC Bhatnagar S “Smart” drug delivery: A window to future of translational medicine” Frontiers in Chemistry 2023 10 1095598 10.3389/fchem.2022.1095598 36688039 PMC 9846181 · doi ↗ · pubmed ↗
- 4Zhang H Fan T Chen W Li Y Wang B Recent advances of two-dimensional materials in smart drug delivery nano-systems Bioactive Materials 2020 5 4 1071 1086 10.1016/j.bioactmat.2020.06.012 32695937 PMC 7363990 · doi ↗ · pubmed ↗
- 5Xiong F Huang S Gu N Magnetic nanoparticles: recent developments in drug delivery system Drug Development and Industrial Pharmacy 2018 44 5 697 706 10.1080/03639045.2017.1421961 29370711 · doi ↗ · pubmed ↗
- 6Kolosnjaj-Tabi J Gibot L Fourquaux I Golzio M Rols MP Electric field-responsive nanoparticles and electric fields: physical, chemical, biological mechanisms and therapeutic prospects Advanced Drug Delivery Reviews 2019 138 56 67 10.1016/j.addr.2018.10.017 30414494 · doi ↗ · pubmed ↗
- 7Hossen S Hossain MK Basher MK Mia MNH Rahman MT Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review Journal of Advanced Research 2019 15 1 18 10.1016/j.jare.2018.06.005 30581608 PMC 6300464 · doi ↗ · pubmed ↗
- 8Crossen SL Goswami T Nanoparticulate carriers for drug delivery Journal of Pharmaceutical and Biopharmaceutical Research 2022 4 1 237 10.25082/JPBR.2022.01.001 · doi ↗