Photocatalytic Addition of N‑Oxazolidinone Radicals to Arenes and Heteroarenes in Batch and in Flow Mode
Sara Ferrario, Sergio Rossi, Niccolò Intini, Julia Bruno-Colmenarez, Marcus Baumann, Maurizio Benaglia

TL;DR
This paper introduces a new photocatalytic method to add N-oxazolidinone radicals to arenes and heteroarenes, offering a sustainable and scalable approach for organic synthesis.
Contribution
The novel generation of N-oxazolidinone radicals via visible light enables efficient C–H functionalization in both batch and flow modes.
Findings
N-oxazolidinone radicals were successfully generated using visible light irradiation.
The C–H functionalization process is efficient, sustainable, and scalable.
Continuous flow processing provides advantages over traditional batch methods.
Abstract
Recent progress in N-centered radical chemistry has paved the way for unprecedented transformations in organic synthesis, particularly through photocatalytic C–N bond formation reactions. Here, unexplored N-oxazolidinone radicals are generated through visible light irradiation, enabling a novel, efficient, sustainable, and scalable C–H functionalization process of arenes and heteroarenes. Crucially, the use of continuous flow processing was found to provide further advantages over standard batch conditions, which will enable further applications of this important transformation.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
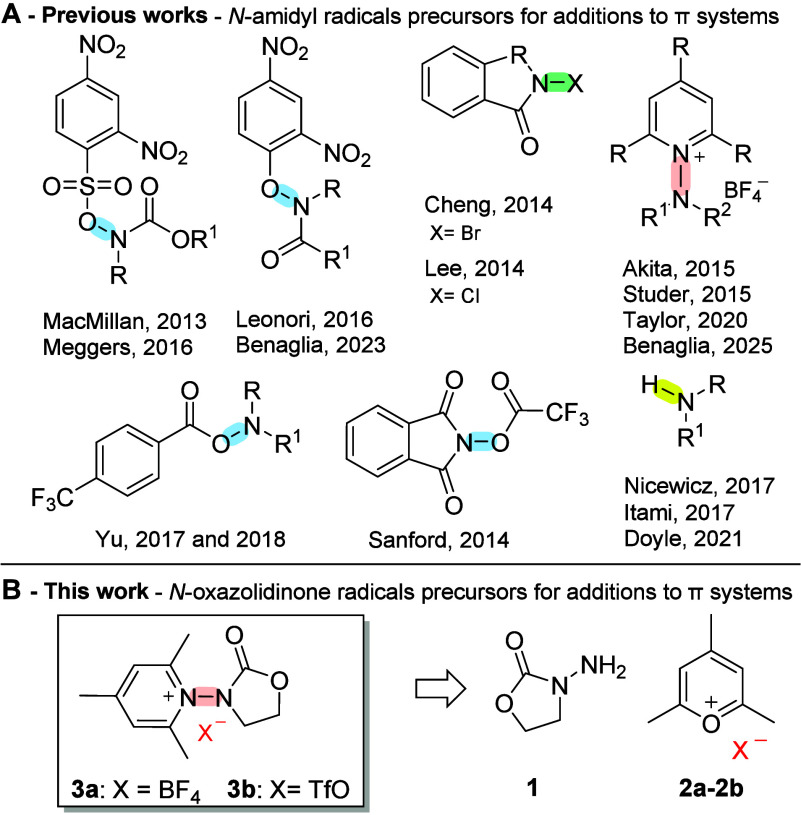 Figure 1
Figure 1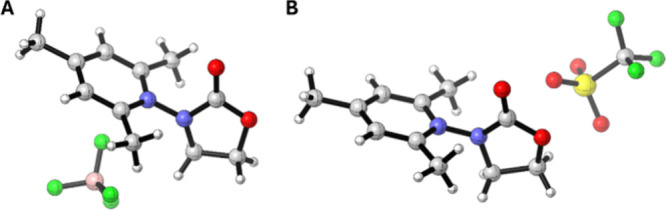 Figure 2
Figure 2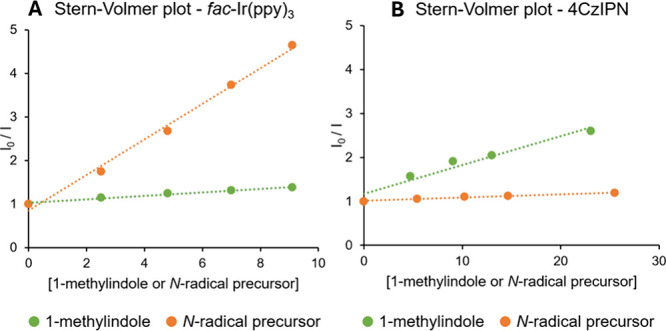 Figure 3
Figure 3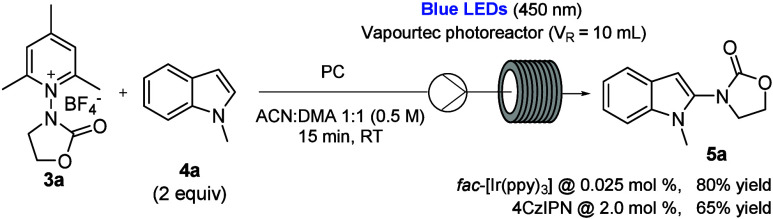 Figure 4
Figure 4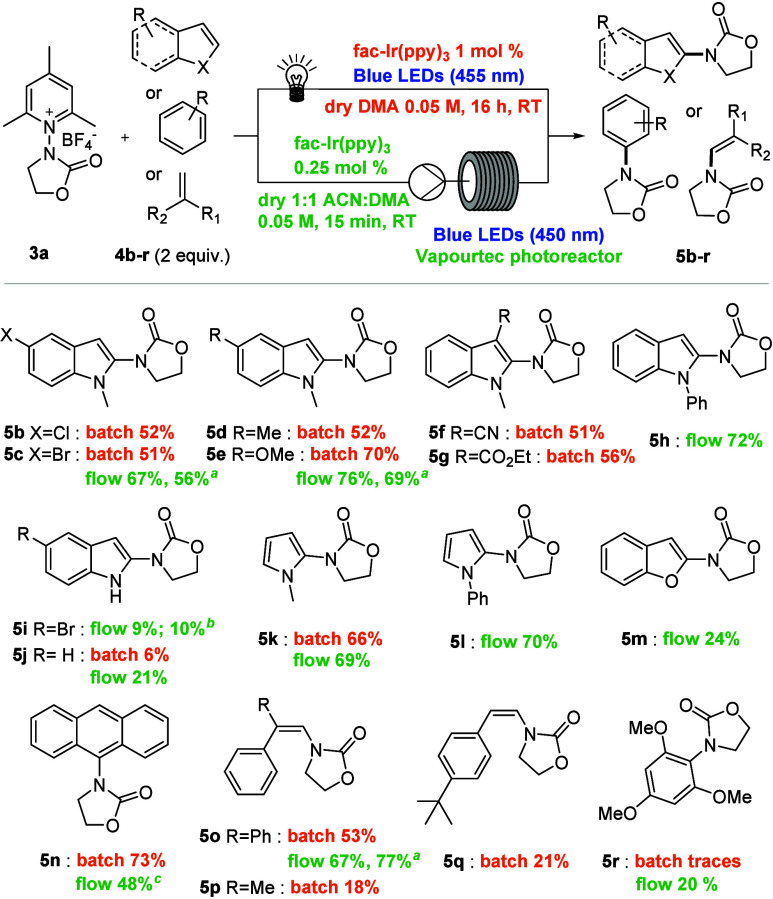 Figure 5
Figure 5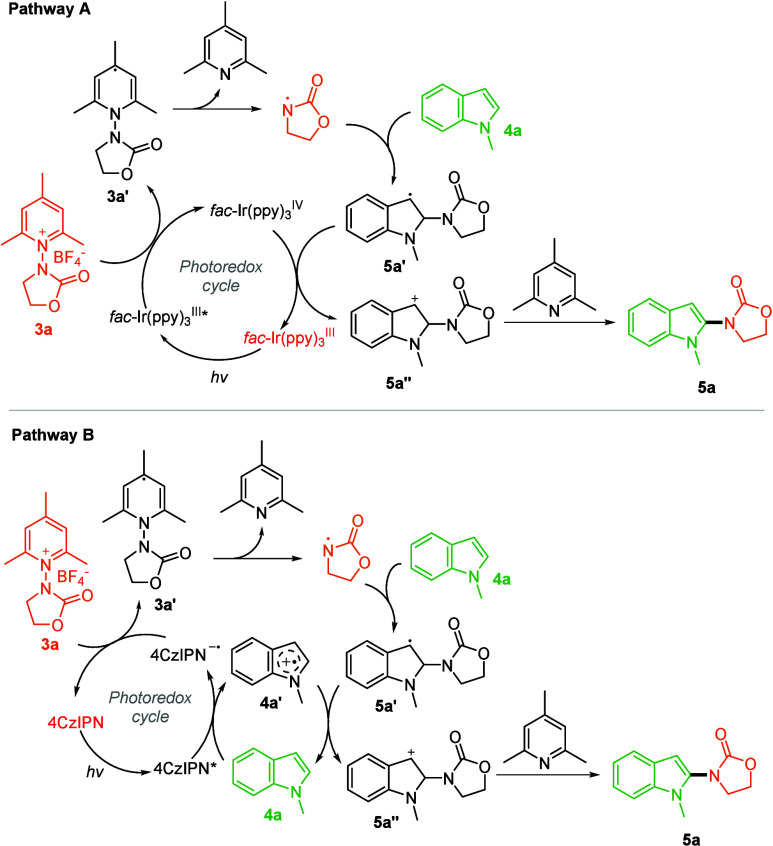 Figure 6
Figure 6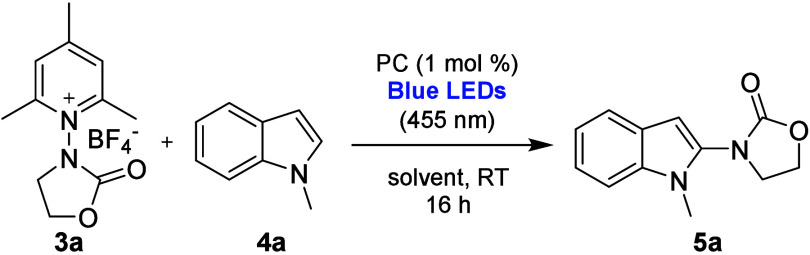 Figure 7
Figure 7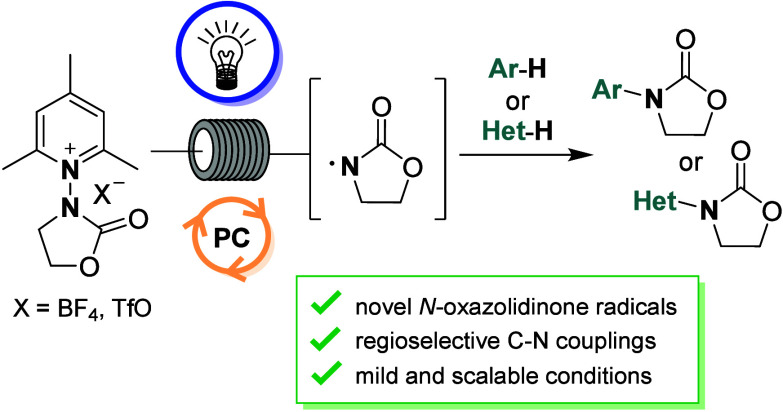 Figure 8
Figure 8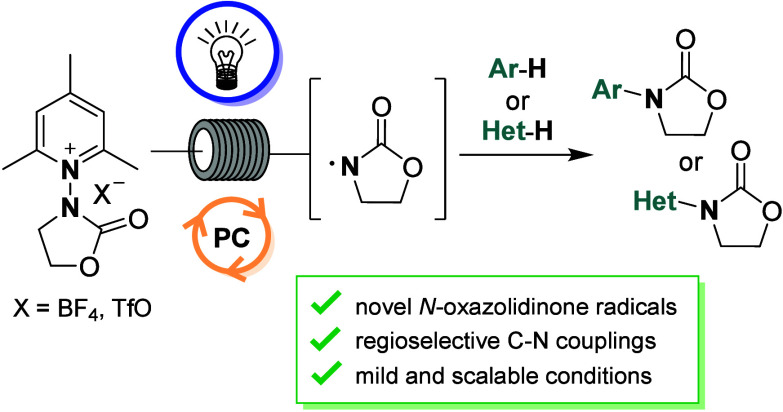 Figure 9
Figure 9- —NextGenerationEU10.13039/100031478
- —Ministero dell?Istruzione, dell?Universit? e della Ricerca10.13039/501100003407
- —Research IrelandNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRadical Photochemical Reactions · Sulfur-Based Synthesis Techniques · Fluorine in Organic Chemistry
Traditional approaches for the formation of C–N bonds typically involve Pd-catalyzed Buchwald–Hartwig amination or Cu-catalyzed Ullmann-type coupling reactions. However, high temperatures and prefunctionalized coupling partners are often required to successfully provide the desired results. ?,? In recent years the chemistry of radicals has been extensively advanced and may offer viable alternatives; for example, N-centered radicals (NCRs) can be prepared and introduced onto the appropriate substrates under mild conditions via photoredox catalysis.?
In this context, the use of metal complexes, such as ruthenium- or iridium-based compounds with polypyridyl ligands or more green and easily synthesizable organic dyes such as 4CzIPN, ?−? ? can promote C–N bond formation reactions under visible-light irradiation. ?−? ? However, reactions involving nitrogen radicals may proceed not only through photocatalytic cycles but also by perpetuating radical propagation processes.?
Among NCRs, amidyl radicals are very versatile species due to their electrophilic chemical behavior and the assumed π-type configuration, resulting from the localization of the unpaired electron in a p orbital (i.e., perpendicular to the nitrogen substituents). Different strategies for the photochemical generation of these reactive species from their precursors are available, such as homolytic cleavage, reductive or oxidative quenching cycles or proton-coupled electron transfer (PCET), all promoted by light. ?,? Specifically, a wide variety of N-amidyl radical precursors have been recently developed (FigureA) involving the cleavage of bonds such as N–O, ?−? ? ? ? ? ? N–halogen, ?,? N–N, ?−? ? ? and N–H. ?−? ?
Furthermore, N-centered radicals, when combined with flow chemistry, are also effective in scaled-up processes, overcoming issues related, for example, to the limited light penetration associated with analogous batch processes. ?,?,?,? As a result, flow technology offers many clear advantages over batch chemistry in terms of reproducibility, safety, and scale-up. ?−? ? ?
In this context, we envisioned the possibility to exploit visible-light photoredox catalysis for the functionalization of different arenes and heteroarenes, exploiting pyridinium salts, often referred to as Katritzky salts,? as radical precursors for the generation of unprecedented N-oxazolidinone radicals (FigureB).
First, a straightforward and convenient protocol to prepare two different novel N-oxazolidinone radical precursors, 2,4,6-trimethyl-1-(2-oxooxazolidin-3-yl)pyridinium tetrafluoroborate 3a and triflate salt 3b, from 1 and 2a,b was identified. Cyclic voltammogram analysis of pyridinium salts 3a and 3b was performed, and the irreversible reduction potentials were determined to be −1.04 and −1.13 V vs Ag/AgCl, respectively (see SI), in agreement with the values of known pyridinium salts. ?,?
Then, we started to study the photocatalytic activation (by N–N bond cleavage) of compound 3a (and 3b) in a model reaction ?,?,? under a traditional batch approach (Table). The generation and regioselective addition of the N-oxazolidinone radical to 1-methylindole 4a occurs via photoredox catalysis, involving blue light and different set-ups, including cylinder- (setup #1, Figure S1) and plate-based (setup #2, Figure S2) photoreactors. First, various photocatalysts (entries 1–3) and solvents (entries 4–7) were evaluated for the reaction optimization, discovering that reactions performed better in polar solvents such as DMA, DMSO, and DMF. DMA was selected due to its more feasible removal through distillation. Then, the photocatalyst loading (entry 8) and the stoichiometry of the reagents (entries 9 and 12) were evaluated. Different photoreactors were also tested, with the plate setup being identified as the most effective (entry 10). The concentration of the reaction was also varied, showing that a lower concentration of 50 mM gave better outcomes (entry 11). In the end, the reaction time was confirmed to be 16 h, in accordance with the lower yield at 8 h (entry 13). The reaction performed with the pyridinium triflate salt 3b afforded the desired compound in 70% yield (entry 14), comparable to the yield of tetrafluoroborate salt 3a (74%, entry 11).
Taking advantage of continuous flow photochemistry, the C–H functionalization of arenes and heteroarenes with the novel N-oxazolidinone radical species was next investigated under flow conditions, using a Vaportec E-Series photochemical reactor (Figure S3). A second round of optimization of the reaction conditions was then necessary, and the stoichiometry, the residence time, as and the nature and the loading of the photocatalyst were investigated. Notably, after a further evaluation of solvents and concentration, a 1:1 mixture of ACN:DMA was identified as good compromise between the experimental setup and reaction conditions, allowing operation with a higher concentration (0.5 M instead of 0.05 M, see SI). From these optimizations, it was found that fac-Ir(ppy)3 photocatalyst showed a better chemical efficiency compared to 4CzIPN; by reacting 1 equiv of 3a with 2 equiv of 4a dissolved in a 1:1 mixture of ACN:DMA as solvent under blue light irradiation for 15 min as the residence time, it was possible to isolate 5a in 80% yield (Scheme). In the same reaction conditions, it was necessary to employ a 2 mol % loading of 4CzIPN to obtain the desired product 5a in 65% yield. Nevertheless, since the organic dye 4CzIPN can be considered a greener alternative to iridium-based photocatalysts, it was effectively used in the reaction scope as well (for a series of control experiments of the reaction in batch and in flow, and their comments, see Table S15).
Having identified the optimized conditions, we started to investigate the scope of the reaction with different arenes and heteroarenes, under both batch and flow conditions (Scheme).
It was found that variously functionalized indoles bearing an alkyl or aryl system with electron-donating or electron-withdrawing groups yielded the products in good to excellent yields (5b–h). It is worth noting that successful results obtained with 3-substituted indoles may pave the way for the derivatization of tryptophan analogs. The reaction with substituted indoles at position 1 resulted in lower yields (5i and 5j). Notably, heteroarenes, such as functionalized pyrroles and benzofuran, turned out to be suitable substrates for this photocatalytic reaction (5k–m). The applicability of the method can also be extended to aromatic compounds, such as anthracene (5n), functionalized styrenes (5o–q), and 1,3,5-trimethoxybenzene (5r), where the reaction under flow conditions performed much better, likely due to the enhanced light penetration.
The scalability of the process under flow conditions was then evaluated. A gram scale reaction (3a, 8 mmol scale) led to the isolation of 1.28 g of product 5a (yield: 74%, with a productivity of 3.2 g·h^–1^, see SI, section 6). To compare the batch and flow approaches, a kinetic study of the photocatalytic reaction was conducted showing that the performance of the optimized continuous flow process is ca. 4.4 times higher than that in batch (Tables S16–17 and Figure S14).
To better understand the photocatalytic process, single crystal X-ray diffraction structures of the N-radical precursors were recorded. The N–N bond length for compounds 3a (FigureA) and 3b (FigureB) was measured to be 1.393 Å. Additionally, the torsional angle around the N–N bonds is 74.93° for tetrafluoroborate salt 3a and 75.49° for triflate salt 3b. As a result, the modest energy associated with the labile N–N bond upon photoactivation may be due to the minimal overlap of the π orbitals of the nitrogen atoms.
Moreover, it was established that the presence of oxygen is not relevant for the process (SI, section 7), which allowed us to investigate the effect of each reaction component on the photocatalyst photoluminescence. In particular, Stern–Volmer analyses were performed by preparing a solution of each photocatalyst, fac-Ir(ppy)3, and 4CzIPN (10^–4^ M) and separately increasing the concentration of the other reaction component, 1-methylindole 4a or N-radical precursor 3a. The photoluminescence lifetime of both photocatalysts linearly decreased with increasing concentrations of both the reagents, as all the corresponding Stern–Volmer plot graphs confirm (Figures S15–S18), but showed an opposite quenching efficiency trend. In more detail, in accordance with the dynamic (or collisional) quenching of fluorescence described by the Stern–Volmer equation, ?,? four Stern–Volmer quenching constants were calculated. Thus, precursor 3a quenches fac-Ir(ppy)3 ten times faster (K D = 4.09 × 10^–1^ M^–1^) than 1-methylindole 4a (K_D_ = 4.04 × 10^–2^ M^–1^) (FigureA). On the contrary, the quenching efficiency of the N-radical precursor 3a on 4CzIPN is significantly lower (K D = 7.20 × 10^–3^ M^–1^) than that of 1-methylindole 4a (K D = 6.59 × 10^–2^ M^–1^) (FigureB). Therefore, two different mechanisms are proposed for the regioselective photocatalytic addition of N-oxazolidinone radicals to 1-methylindole (Scheme). In the first proposed reaction mechanism (Scheme, Pathway A), once a photon is absorbed by the fac-tris(2-phenylpyridine)iridium(III) photocatalyst (E red ^0^ Ir(III)*/Ir(IV) ∼ −1.73 V vs SCE),? the excited species reduces the pyridinium salt 3a via single electron transfer (SET). The fragmentation of precursor intermediate 3a′ releases the N-oxazolidinone radical and 2,4,6-collidine. The regioselective addition of the radical to the α-position of 1-methylindole provides radical intermediate 5a′, which is then oxidized via single electron transfer, while the photocatalyst is regenerated. The aminated indole 5a is formed by deprotonation of 5a″, since 2,4,6-collidine acts as a base and allows the catalytic cycle.
In contrast, in the second proposed mechanism (Scheme, Pathway B), the excited 4CzIPN (E red ^*^ = +1.35 V vs SCE)? oxidizes 1-methylindole 4a via SET.? The 4CzIPN radical anion reduces the N-radical precursor 3a, while the photocatalyst is regenerated. The fragmentation of the precursor intermediate 3a′ provides 2,4,6-collidine and generates the N-oxazolidinone radical. The oxazolidinone radical reacts at the α-position of 1-methylindole 4a to generate the radical intermediate 5a′, which is subsequently oxidized to 5a″ upon SET. Lastly, the deprotonation of 5a″ yields the desired amidated indole 5a.
In conclusion, a straightforward protocol was developed for the synthesis of a novel class of N-radical precursors. The photocatalytic addition of the N-oxazolidinone radical to arenes and heteroarenes was successfully performed under batch and continuous flow conditions, obtaining the desired products in high yields (up to 80%). In addition, the scalability of the process was confirmed by performing a gram-scale reaction, with a good productivity and STY under flow conditions (respectively, 3.2 g·h^–1^ and 200 g·h^–1^·L^–1^). Furthermore, single crystal structures, cyclic voltammetry, and photophysical studies enabled a detailed characterization of the nitrogen radical precursors and a better understanding of the reaction. In the end, two different mechanisms are proposed for the two different photocatalysts used in the transformation.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pratley C.Fenner S.Murphy J. A.Nitrogen-Centered Radicals in Functionalization of sp 2 Systems: Generation, Reactivity, and Applications in Synthesis Chem. Rev.20221228181826010.1021/acs.chemrev.1c 0083135285636 · doi ↗ · pubmed ↗
- 2Kärkäs M. D.Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications ACS Catal.201774999502210.1021/acscatal.7b 01385 · doi ↗
- 3Chan A. Y.Perry I. B.Bissonnette N. B.Buksh B. F.Edwards G. A.Frye L. I.Garry O. L.Lavagnino M. N.Li B. X.Liang Y.Mao E.Millet A.Oakley J. V.Reed N. L.Sakai H. A.Seath C. P.Mac Millan D. W. C.Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis Chem. Rev.20221221485154210.1021/acs.chemrev.1c 0038334793128 PMC 12232520 · doi ↗ · pubmed ↗
- 4Crespi S.Fagnoni M.Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy Chem. Rev.20201209790983310.1021/acs.chemrev.0c 0027832786419 PMC 8009483 · doi ↗ · pubmed ↗
- 5Marzo L.Pagire S. K.Reiser O.König B.Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis?Angew. Chem., Int. Ed.201857100341007210.1002/anie.20170976629457971 · doi ↗ · pubmed ↗
- 6Shaw M. H.Twilton J.Mac Millan D. W. C.Photoredox Catalysis in Organic Chemistry J. Org. Chem.2016816898692610.1021/acs.joc.6b 0144927477076 PMC 4994065 · doi ↗ · pubmed ↗
- 7Luo J.Zhang J.Donor-Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp 3)-C(sp 2) Cross-Coupling ACS Catal.2016687387710.1021/acscatal.5b 02204 · doi ↗
- 8Speckmeier E.Fischer T. G.Zeitler K.A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes J. Am. Chem. Soc.2018140153531536510.1021/jacs.8b 0893330277767 · doi ↗ · pubmed ↗