Hereditary Spherocytosis due to an SPTA1 Nonsense Mutation Coinherited With α spectrinLELY in Trans
María‐Angustias Molina‐Arrebola, Barbara J. Bain

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
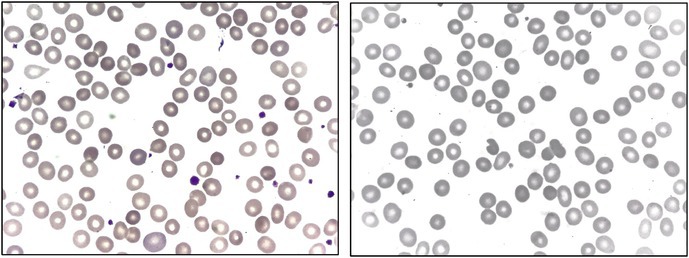 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsErythrocyte Function and Pathophysiology · Blood properties and coagulation · Caveolin-1 and cellular processes
A 50‐year‐old Spanish woman was referred with a diagnosis of hereditary spherocytosis. She had undergone cholecystectomy 8 years earlier and had required blood transfusions during each of her three pregnancies; none of her children was affected. Initial laboratory data showed a hemoglobin concentration of 93 g/L, mean corpuscular volume (MCV) of 95.7 fL, mean corpuscular hemoglobin (MCH) of 33 pg, and mean corpuscular hemoglobin concentration (MCHC) of 354 g/L. The absolute reticulocyte count was elevated at 218 × 10^9^/L. Platelet and leukocyte counts were normal. Her blood film showed moderate numbers of spherocytes and small numbers of other poikilocytes (tear‐drop poikilocytes, bite cells and irregularly contracted cells) (images, May–Grünwald–Giemsa ×100 objective). There was also polychromasia. Total bilirubin was elevated at 2.25 mg/dL, with a direct fraction of 0.48 mg/dL. Haptoglobin was undetectable. Lactate dehydrogenase and liver enzymes were normal. Flow cytometry‐based eosin‐5‐maleimide (EMA) binding assay was repeatedly normal, with a fluorescence ratio of 0.96. Abdominal ultrasonography revealed a large, homogeneous spleen measuring 13.4 cm in its longest diameter.
A next‐generation sequencing (NGS) panel for hereditary hemolytic anemias was analyzed. This showed the nonsense variant c.4519C>T p.(Arg1507*) in the SPTA1 gene, predicting substitution of an arginine by a premature stop codon at position 1507 [1]. The patient also carried two common variants in *SPTA1—*c.6531‐12C>T and c.5572C>G p.(Leu1858Val). In addition, a heterozygous variant of uncertain significance was detected in the GSR gene, which encodes glutathione reductase: c.616A>C p.(Thr206Pro).
Hereditary spherocytosis (HS) comprises a heterogeneous group of red cell membrane disorders, with an estimated prevalence of at least one in 2000 among individuals of Northern European descent. Approximately three‐quarters of cases have an autosomal dominant inheritance pattern, while the remainder are either sporadic de novo mutations or inherited in an autosomal recessive manner [2]. Pathogenic mutation of SPTA1 is responsible in only a minority of cases, occurring when there is homozygosity, compound heterozygosity, or coinheritance with a low expression allele in trans, such as α spectrin^LELY^ (Low Expression Lyon) [3] or α spectrin^LEPRA^ (Low Expression Prague) [4]. The designation α spectrin^LELY^ has sometimes been used to refer specifically to the c.6531‐12C>T variant but often to designate its coexistence with the c.5572C>G p.(Leu1858Val) variant in cis. This polymorphism, as observed in our patient, is common in the general population, with an allele frequency of approximately 20%–30% [4].
EMA binding is often used for confirmation of a diagnosis of hereditary spherocytosis when spherocytes are observed in the blood film of a patient with chronic hemolytic anemia. However, the observation of normal or borderline EMA binding does not exclude this diagnosis, and in this circumstance, detailed genetic analysis is indicated. It is also worth noting that this patient harbored a heterozygous variant in the GSR gene, which encodes glutathione reductase. While its pathogenicity is uncertain, impaired glutathione recycling may reduce the erythrocyte's ability to manage oxidative stress, potentially acting as a phenotype modifier in the setting of chronic hemolysis.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1https://www.ncbi.nlm.nih.gov/clinvar/variation/1343229/?oq=1343229&m=NM_003126.4(SPTA 1):c.4519 C%3ET%20(p.Arg 1507 Ter).
- 2S. Chonat , M. Risinger , H. Sakthivel , et al., “The Spectrum of SPTA 1‐Associated Hereditary Spherocytosis,” Frontiers in Physiology 10 (2019): 815.31333484 10.3389/fphys.2019.00815 PMC 6617536 · doi ↗ · pubmed ↗
- 3R. Wilmotte , J. Marechal , and J. Delaunay , “Mutation at Position −12 of Intron 45 (c → t) Plays a Prevalent Role in the Partial Skipping of Exon 46 From the Transcript of Allele alpha LELY in Erythroid Cells,” British Journal of Haematology 104, no. 4 (1999): 855–859.10192450 10.1046/j.1365-2141.1999.01271.x · doi ↗ · pubmed ↗
- 4J. Delaunay , V. Nouyrigat , A. Proust , et al., “Different Impacts of Alleles α LEPRA and α LELY as Assessed Versus a Novel, Virtually Null Allele of the SPTA 1 Gene in Trans ,” British Journal of Haematology 127 (2004): 118–122.15384986 10.1111/j.1365-2141.2004.05160.x · doi ↗ · pubmed ↗