Tundrisphaera macrotermitis sp. nov., a novel member of the family Isosphaeraceae isolated from the gut of a fungus-growing termite
Nicolai Kallscheuer, Kim-Loreen Carlstedt, Jonathan Hammer, Tom Haufschild, René Benndorf, Z. Wilhelm de Beer, Michael Poulsen, Christine Beemelmanns, Christian Jogler

TL;DR
A new bacterial species, Tundrisphaera macrotermitis, was discovered in the gut of a fungus-growing termite and is described based on its genetic and phenotypic traits.
Contribution
The discovery and characterization of a novel bacterial species from the gut of a fungus-growing termite.
Findings
The isolate TA3T belongs to the family Isosphaeraceae and has distinct genomic and phenotypic features.
Comparative genomics showed significant differences from the closest relative Tundrisphaera lichenicola.
TA3T is a new species in the genus Tundrisphaera, with a genome size of 7.23 Mbp and four plasmids.
Abstract
A pink-pigmented, neutrophilic and mesophilic strain, TA3T, was isolated from the hindgut of a fungus-growing termite of the species Macrotermes natalensis. Phylogenetic analysis placed the strain in the family Isosphaeraceae, order Isosphaerales, class Planctomycetia, phylum Planctomycetota. The isolate turned out to be an aerobic chemoorganoheterotroph capable of growth under microaerobic conditions. Cells are non-motile, spherical, and either form shapeless aggregates or grow as single cells. The average cell size (length x width) is 2.5 ± 0.3 μm x 2.3 ± 0.2 μm. Cells divide asymmetrically by budding. Optimum pH and temperature for growth are 7.5 (range 6.0–9.0) and 24 °C (range 18–28 °C), respectively. The strain has a genome size of 7.23 Mbp with 69.3% DNA G + C content and it contains four plasmids. Since the genome of the currently known closest relative Tundrisphaera lichenicola…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1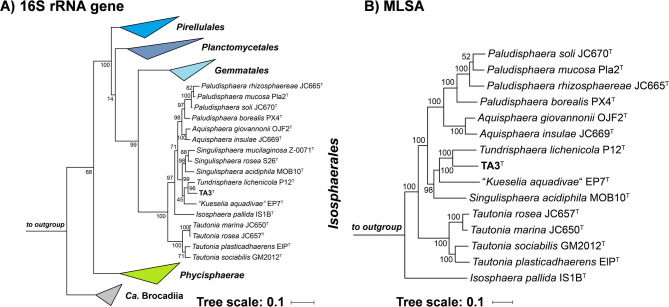 Figure 2
Figure 2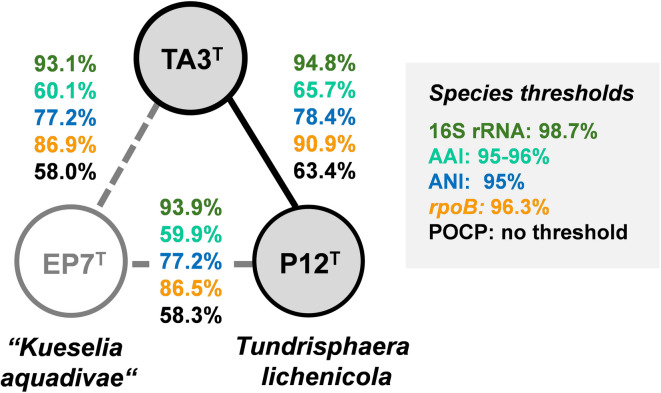 Figure 3
Figure 3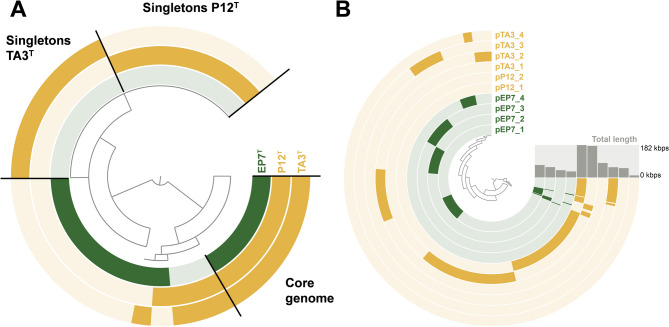 Figure 4
Figure 4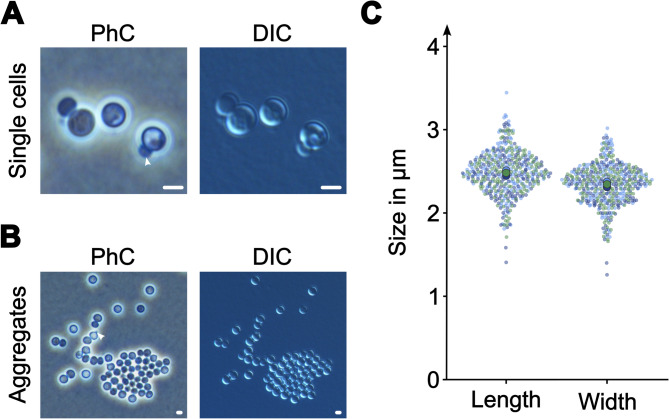 Figure 5
Figure 5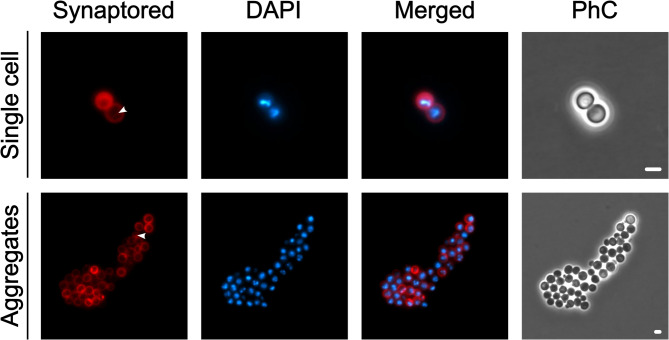 Figure 6
Figure 6- —Friedrich-Schiller-Universität Jena (1010)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect and Arachnid Ecology and Behavior · Genomics and Phylogenetic Studies · Insect symbiosis and bacterial influences
Introduction
The phylum Planctomycetota represents a distinctive group of bacteria known for their unusual cell biology and ecological adaptability^1,2^. Phylum members are recognized for an uncommon cellular architecture including extensive membrane invaginations, condensed DNA and asymmetric cell division by budding^3–6^. Cultivation-dependent and -independent studies have identified members of the two validly described classes Planctomycetia and Phycisphaerae, and the two provisional classes Ca**ndidatus Brocadiia and Candidatus Uabimicrobiia in a range of habitats. These habitats include freshwater^7,8, ^marine^9–12 ^and terrestrial environments^13,14^. In aquatic ecosystems, higher abundances are typically observed on biotic and abiotic surfaces than in the surrounding water^15,16^. This is in line with the finding that planctomycetes are talented degraders of phototroph-derived polysaccharides^17^, which requires physical contact with micro- and macroscopic phototrophs such as cyanobacteria, seagrasses, macroalgae, etc. ^16^. Consequently, planctomycetes play important roles in global biogeochemical cycles in carbon but also nitrogen transformations; the latter in particular with regard to anaerobic ammonium oxidation (anammox) metabolism performed by members of the class Ca. Brocadiia^18^.
Within the well-sudied Planctomycetia^2 the family* Isosphaeraceae* (order Isosphaerales) comprises aerobic to facultatively anaerobic, spherical bacteria capable of thriving in both, hot and cold environments^^19–21^. Most Isosphaeraceae members have been isolated from peat bogs, lichen-dominated soil and limnic environments^8,22^. Polysaccharide degradation capabilities in the family have been investigated both by cultivation studies and comparative genomics^23–26^.
Planctomycetes have also been found in host-associated environments, such as, the gastrointestinal tracts of insects, with relative abundances of up to 33% ^27–29^, though their functional contributions in these niches remain poorly understood. The gut of the fungus-growing termite species Macrotermes natalensis hosts a complex but stable microbial community within which the phylum Planctomycetota is among the six most abundant phyla^30^. However, the isolation and characterization of planctomycetes from termite guts also remain sparse, leaving gaps in our understanding of potential adaptations to host-associated lifestyles.
Here, we describe strain TA3^T^ that was isolated from the hindgut of M. natalensis after cultivation under aerobic conditions for several months. The isolate belongs to the family Isosphaeraceae (phylum Planctomycetota), which currently consists of 16 described species in seven genera. The closest related known species, Tundrisphaera lichenicola, is constituted by two strains that were isolated from peatland or tundra soil in Russia^22^. Their genomes have not been sequenced yet.
The characterization of strain TA3 is accompanied by genome sequencing of the type strain of T. lichenicola. The determination of the genome sequence of the previously isolated strain allowed for more detailed phylogenetic analyses, including whole genome-based markers that, together with differences in phenotypic characteristics, support the delineation of strain TA3 from the other described species in the genus Tundrisphaera.
Materials and methods
Sampling, strain isolation and sequencing of the 16 S rRNA gene
Termites of the species M. natalensis were collected from the University of Pretoria Experimental Farm in Pretoria, South Africa in 2016 (latitude − 25.743028, longitude 28.260972) (Fig. 1). Termite specimens were transferred to the laboratory and stored in 50% glycerol. Termites were dissected and split into five parts: crop, foregut, midgut, hindgut, and the remaining termite body. Individual parts were transferred into 1.5 mL reaction tubes with 200 µL of autoclaved water and antifungals (50 µg/mL cycloheximide and 100 µg/mL nystatin). After 24 h of incubation at 4 °C in presence of the antifungals, 10 µL of each sample was transferred to limnic M1 agar supplemented with 200 mg/L ampicillin and 500 mg/L streptomycin prepared as previously described^31^. The plates were incubated under aerobic conditions at 18 °C in plastic boxes. Plates were sealed with Parafilm and incubated with wet papers towels to avoid that plates dry out during longer incubation periods. After five months of incubation, a pinkish colony was visible on the plate inoculated with the hindgut sample. The pinkish colony, designated strain TA3^T^, was re-streaked on fresh plates and was maintained in liquid limnic M1 medium without antibiotics. The 16S rRNA gene of strain TA3^T^ was amplified by PCR, purified based on a standardized workflow^32^ and sequenced at Macrogen Europe (Amsterdam, The Netherlands).
Fig. 1. Isolation of strain TA3^T^ from a* Macrotermes natalensis* colony. (A) Termite workers use a mixture of soil and saliva to construct walls and mounts that serve to protect the colony. (B) Termite workers dedicated to the welfare of the queen, transporting eggs and feeding her. (C) Digestive tract from which strain TA3^T^ was isolated. The hindgut is indicated by the white arrow.
Physiological analyses
For determination of the temperature optimum for growth, 100 µL supernatant of an exponentially growing culture were plated and plates were incubated in triplicates at temperatures ranging from 4 to 37 °C. Plates were checked daily, and growth was evaluated by the time required until visible colonies were formed. The temperature at which colonies appeared the earliest was considered the temperature optimum for growth. The pH optimum for growth was determined in liquid cultures of limnic M3 medium (same recipe as limnic M1 medium but with amounts of peptone and yeast extract increased to 1 g/L each). As buffering agents, the medium contained either 100 mM of 2-(N-morpholino)ethanesulfonic acid (MES) for pH 5.0 and 6.0, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) for pH 7.0 and 8.0 or N-cyclohexyl-2-aminoethanesulfonic acid (CHES) for pH 9.0 and 10.0. Growth was evaluated by measuring the optical density at 600 nm (OD_600_). Growth under anoxic conditions was examined using solidified and liquid limnic M3 medium. The medium was anaerobized by flushing it with nitrogen gas for two minutes until oxygen concentrations below 1 µmol/L were achieved. Plates were then transferred to an anaerobic jar (nox-18, SY-LAB, Neupurkersdorf, Austria). Plates and sealed serum bottles were incubated at 24 °C.
Microscopy and cell size determination
For cell size determination and fluorescence microscopy, a Nikon Eclipse Ti2 inverse microscope with two different set-ups was used as described earlier^33^. To analyze cell size, cells of strain TA3^T^ were handled and images were analyzed as previously described^31^. Briefly, cells grown in limnic M3 medium were mounted on a 1% (w/v) agarose cushion dissolved in the same medium. Images were loaded into FIJI^34^, all three RGB-channels were combined and tiff files were transferred to BacStalk^35^. Segmentation settings were adjusted to 25 and 15 pixels for cell size and minimum cell size, respectively. Segmented cells (three replicates with 150 cells each) were checked manually for detection errors. The data was transferred to SuperPlotsOfData^36^. For visualization purposes, brightness and contrast were adjusted manually. For fluorescence microscopy, 500 µL of a culture was stained with 3 µL 4′,6-diamidin-2-phenylindole (DAPI) and 1 µL Synaptored for 30 min. Cells were washed 3 times with limnic M3 medium. After immobilization on a 1% (w/v) agarose pad dissolved in the same medium, cells were imaged with a DAPI filter (Semrock; DAPI-1160B) and a Texas Red longpass filter (AHF; 560/40x, 600 DC, 610 LP). Images were transferred to FIJI (if necessary, images were merged with the in-FIJI function) and brightness and contrast were adjusted manually to enhance visibility.
Genomic DNA isolation, genome sequencing, annotation and analysis
Isolation of genomic DNA as well as genome sequencing, assembly and polishing of strain TA3^T^ were performed as previously described^31^. The genome was assembled de novo from Oxford Nanopore long reads and polished with Illumina short reads. Illumina sequencing was performed by Eurofins Genomics (Ebersberg, Germany). T. lichenicola P12^T^ was ordered from the Belgian culture collection BCCM/LMG (deposition number LMG 29571) and processed in the same way as strain TA3^T^. The final genome sequences were checked for completeness using BUSCO v5.8.2. Coding density and DNA G + C content were analyzed with checkM v1.2.3. After annotation with prokka v1.14.5, the chromosome was rotated to the start codon of the replication initiator protein-encoding gene dnaA and was re-annotated with PGAP version 2025-05-06.build7983. Plasmids were rotated to the start codon of the chromosome partitioning ATPase-encoding gene parA when such a protein was annotated.
Nucleotide sequence accession numbers
The 16S rRNA gene sequences of strain TA3^T^ and T. lichenicola P12^T^ are available from the NCBI GenBank database under the accession numbers PV747909 and PV748000, respectively. Genome sequence information for strain TA3^T^ was deposited under the accession numbers CP195100 (chromosome), CP195101 (pTA3_1), CP195102 (pTA3_2), CP195103 (pTA3_3) and CP195104 (pTA3_4). Genome sequence information for T. lichenicola P12^T^ was deposited under the accession numbers CP195106 (chromosome), CP195107 (pP12_1) and CP195108 (pP12_2).
Phylogenetic and genome-based analyses
The 16S rRNA gene sequence of strain TA3^T^ was extracted from the prokka-annotated genome and was used for the identification of the current closest relatives using NCBI BLAST^37^. A maximum-likelihood 16S rRNA gene sequence-based phylogeny was computed for the novel strain and the described type strains of all species in the current phylum Planctomycetota (as of June 2025). Three sequences from strains outside of the phylum Planctomycetota, but part of the Planctomycetota-Verrucomicrobiota-Chlamydiota (PVC) superphylum, namely Opitutus terrae (NCBI acc. no. AJ229235), Kiritimatiella glycovorans (acc. no. NR_146840) and Lentisphaera araneosa (accession number NR_027571), served as outgroup. The alignment of the 16S rRNA gene sequences was performed with ClustalW^38^ and FastTree v2.2 was used for tree reconstruction with 1000 bootstrap replications^39^. A multi-locus sequence analysis (MLSA)-based phylogeny was performed using autoMLST with 500 bootstrap replicates^40^. The analysis included the genomes of all described members of the family Isospheraceae and genomes of Rhodopirellula baltica SH1^T^ (GenBank acc. no. BX119912.1), Pirellula staleyi DSM 6068^T^ (acc. no. CP001848.1) and Blastopirellula marina DSM 3645^T^ (acc. no. GCA_000153105.1) (all belonging to the family Pirellulaceae) served as outgroup. Phylogenetic trees were visualized with iTOL v6 ^41^. The 16S rRNA gene sequence similarity matrix was obtained with TaxonDC^42^ based on the ClustalW alignment that was also used for the construction of the phylogenetic tree. Average amino acid identities (AAI) and average nucleotide identities (ANI) were calculated using respective scripts of the enveomics collection^43^. Additional phylogenetic markers, i.e. rpoB sequence similarity and percentage of conserved proteins (POCP), were calculated as described^44,45^. Digital DNA-DNA hybridization (dDDH) values were calculated with the Genome-to-Genome Distance Calculator of the Type Strain Genome Server (TYGS) (https://ggdc.dsmz.de). The pangenome of selected strains was generated with anvi’o v.8 ^46^. Biosynthetic gene clusters (BGCs) were predicted with antiSMASH v.8.0 ^47^ and carbohydrate-active enzymes (CAZymes) with dbCAN3^48^. The presence of phage genes was analyzed with PHASTEST^49^. The clustering of protein sequences encoded by plasmid-located genes was performed with CLANS from the MPI Bioinformatics Toolkit^50^.
Results and discussion
Phylogenetic inference
To estimate the phylogenetic position of the novel strain TA3^T^, its 1508 bp 16S rRNA gene sequence was compared against NCBI’s core nucleotide database using blastn. The analysis yielded highest similarities in the range of 93–95% to two members of the family Isosphaeraceae, T. lichenicola P12^T^ and the recently described strain “Kueselia aquadivae” EP7^T^ ^22,51^. Since the genome of T. lichenicola P12^T^ had not been sequenced, the strain was ordered from the Belgian collection BCCM/LMG (deposition number LMG 29571), and its genome sequence was determined along with that of the novel isolate. This allowed for more accurate phylogenetic analyses based on genome-based markers including AAI, ANI, POCP and sequence similarity of a ca. 1300 bp partial sequence of the rpoB gene encoding the β-subunit of RNA polymerase. Maximum-likelihood phylogenetic trees based on 16S rRNA gene sequences and MLSA (Fig. 2) substantiated the suspected clustering in proximity to the two mentioned relatives, identifying T. lichenicola P12^T^ as the currently closest characterized relative of strain TA3^T^. The exact sequence similarity of 94.8% based on the full-length 16S rRNA genes of strains P12^T^ and TA3^T^ extracted from the genome fell above the accepted genus threshold of 94.5%, but below the species threshold of 98.7% ^52^. The inference that strain TA3^T^ is a novel species in the genus Tundrisphaera was supported by the analysis of whole genome-based phylogenetic markers (Fig. 3). The partial rpoB sequence similarity of 96.3% obtained during comparison of strains TA3^T^ and P12^T^ is the exact value of the proposed species threshold^44^, whereas values for ANI and AAI were well below the accepted threshold for species delineation^53,54^. While POCP values are typically only used for the delineation of genera (genus threshold 50%) ^45^, the four phylogenetic markers with available species thresholds (16S rRNA gene sequence similarity, ANI, AAI and partial rpoB sequence similarity) support the position of TA3^T^ as member of a novel species.
Fig. 2. Phylogenetic placement. (A) Maximum likelihood phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship of strain TA3^T^ and the characterized members in the order Isosphaerales. FastTree was used for tree reconstruction with 1000 bootstrap replications (given at the nodes, in %) and Opitutus terrae (NCBI acc. no. AJ229235), Kiritimatiella glycovorans (acc. no. NR_146840) and Lentisphaera araneosa (acc. no. NR_027571), served as outgroup. Bar, 0.1 nucleotide substitutions per position. (B) Multi-locus sequence analysis (MLSA)-based phylogenetic tree based on the characterized members in the order Isosphaerales. The tree was computed based on a set of at least 30 single-copy gene-encoded proteins in a maximum likelihood approach with 500 bootstrap replications using the tool autoMLST (see Materials and Methods section for details). Bootstrap values are given at the nodes. Bar, 0.1 amino acid substitution per position. Phylogenetic trees were visualized with iTOL v6.
Fig. 3. Comparison of phylogenetic markers for species delineation. Markers used: 16S rRNA gene sequence identity (16S rRNA), average amino acid identity (AAI), average nucleotide identity (ANI), sequence similarity of a partial sequence of the rpoB gene (rpoB), percentage of conserved proteins (POCP), digital DNA-DNA hybridization (dDDH). Grey-filled circles and full lines indicate a relationship on the genus level.
Comparison of genomic features
The genome of strain TA3^T^ has a size of 7.23 Mbp. It comprises the chromosome and four plasmids. The presence of one to five plasmids is a common feature of the order Isosphaerales in comparison to the other orders in the class Planctomycetia in which plasmid occurrence in characterized strains is sporadic^23,55^. With 7.80 Mbp, the genome of T. lichenicola P12^T^ is slightly larger, but the strain only contains two plasmids. Both genomes have high DNA G + C content that differs by nearly 5 percentage points (Table 1). Strain TA3^T^ has five copies each of 5S, 16S and 23S rRNA genes that are arranged in five rrn operons (16S–23S-5S rRNA genes) whereas T. lichenicola P12^T^ has four. While the organization of rRNA genes in operons is common in most bacteria, many planctomycetotal species have “unlinked” rRNA genes^56,57^. The numbers of tRNAs, coding sequences per Mbp and coding densities of the three genomes are comparable. Relative numbers of 26–28% of hypothetical protein-encoding genes are in the expected range for planctomycetotal genomes which are among the bacterial genomes with the highest numbers of hypothetical proteins^58^.
Table 1. Comparison of phenotypic features.CharacteristicsTA3^T^Tundrisphaera lichenicola P12^T^“Kueselia**aquadivae” EP7^T^Sampling InformationLocationExperimental termite farm, Pretoria (South Africa)Nadym Region, Yamalo-Nenets Autonomous Okrug (Russia)Hainich National Park, Thuringia (Germany)Sampled MaterialHindgut of Macrotermes natalensisOxic Layer of PeatPercolate of Drainage CollectorPhenotypic featuresPigmentationpinkpinklight pinkCell ShapesphericalsphericalsphericalSize (Length x width) (µm)2.5 × 2.32.2-3.02.4 × 2.2Cell Division Modeasymmetric (polar) cell division (budding)asymmetric (polar) cell division (budding)asymmetric (polar) cell division (budding)Temperature Range (optimum) (°C)18–28 (24)4–28 (15–22)10–25 (18–21)pH Range (optimum)6.0–9.0 (7.5)4.5–6.8 (5.5–6.0)6.0–9.0 (7.5)Relation to Oxygenaerobic, growth under microaerobic conditionsstrictly aerobicfacultatively anaerobicMotilitynon-motilenon-motilenon-motileStalksnot observedHoldfast-like appendagesnot observedAggregatesshort chains, or shapeless aggregatesshort chainssmall, microscopic aggregates
Pangenome reconstruction, analysis of secondary metabolite-associated biosynthetic gene clusters and carbohydrate-active enzymes
The constructed pangenome based on the genomes of “K. aquadivae” EP7^T^ and the two sequenced Tundrisphaera spp. consisted of 11,030 clusters, of which 2,078 were present in all three genomes (core genome). The analysis yielded 2,310 singleton genes for strain TA3^T^ and 2,544 for T. lichenicola P12^T^ (Fig. 4A). In silico mining of planctomycetotal genomes using antiSMASH is typically performed to get an overview on secondary metabolite biosynthetic capabilities of individual strains and to identify potential talented producers of novel bioactive compounds^4,59,60^. As was also the case for the three strains compared here (Table 2), antiSMASH yielded around one BGC per Mbp for planctomycetotal genomes. The analysis yielded a conserved set of four terpenoid biosynthesis-related BGCs, two type I and one type III polyketide synthase clusters across all three genomes and 1–2 additional strain/species-specific clusters. The latter were predicted to be involved in the biosynthesis of acyl amino acids or ribosomally and non-ribosomally synthesized peptides. AntiSMASH predicted an additional non-ribosomal peptide synthetase-encoding gene in strain TA3^T^ that was absent in the other two genomes.
Fig. 4. Pangenome and ‘panplasmidome’ analyses. Each open circle represents the pangenome (A) or ‘panplasmidome’ (B) of all genomes or plasmids, with darker shading indicating the presence of a given gene in the corresponding genome or plasmid. A) The core genome and singletons of the two Tundrisphaera species are highlighted. B) ‘Total length’ denotes the combined length of the analyzed plasmids, expressed in kilobase pairs (kbps).
Table 2. Comparison of genomic and genome-encoded features.CharacteristicsTA3^T^Tundrisphaera lichenicola P12^T^“Kueselia**aquadivae” EP7^T^Genomic featuresGenome size (bp)7,233,8707,799,4497,199,273Plasmids424DNA G + C (%)69.364.766.7Genes5,8626,2005,685Genes/Mbp810795790Protein-coding genes5,7506,0575,563Protein-coding genes/Mbp795777773Hypothetical proteins1,5401,5531,534Hypothetical proteins (%)26.825.627.6Coding density (%)88.186.886.3rRNA genes (5S–16S-23S)5,5,54,4,44,4,4tRNA genes645158Secondary metabolite-associated biosynthetic gene clustersTerpene444Type I polyketide synthase223Type III polyketide synthase111Ribosomally synthesized modified peptide210Non-ribosomal peptide synthetase100Acyl amino acid020BGCs per Mbp1.41.31.1Carbohydrate-active enzymesGlycoside hydrolases (GH)585055Glycosyltransferases (GT)909985Polysaccharide lyases (PL)303Carbohydrate esterases (CE)192422Carbohydrate-bind. modules (CBM)162312Auxiliary activities (AA)452CAZyme genes190201179CAZyme genes per Mbp262625Based on the PGAP-annotated genomes
The number of putative CAZyme-encoding genes identified in the three genomes fell in the range of 180–200 with nearly identical values of 25–26 CAZyme genes per Mbp. The sampling spots from which strains TA3^T^ and P12^T^ were isolated differ in several properties, including temperature, humidity, and host association. Hence, we checked whether habitat-specific requirements for the degradation of carbohydrates were reflected in differences in their CAZyme portfolios. However, this revealed only minor differences for the presence/absence of the different subfamilies. In a comparison of strains P12^T^ and TA3^T^, eleven subfamilies were specific for P12^T^ (AA1, CBM47, CBM57, CBM66, CBM67, CE4, GH1, GH5, GH51, GH141, GT1) while tnine subfamilies were specific for TA3^T^ (CBM2, GH24, GH26, GH76, GH123, GH140, GT28, PL10, PL11). Striking was the lack of putative polysaccharide lyases (PLs) in T. lichenicola P12^T^ and the presence of different PL subfamilies with partly overlapping specificities in the other two strains (PL10 and PL11 in strain TA3^T^ and PL9 and PL42 in “K. aquadivae” EP7^T^). PL9 and PL11 are presumably involved in the degradation of rhamnogalacturonan, PL10 in the degradation of pectate and PL42 of L-rhamnose-α-1,4-D-glucuronate.
Interestingly, none of the TA3^T^-specific CAZyme subfamilies was previously found to be termite specific^61^. This finding along with the large genome of strain TA3^T^ questions if this strain is indeed among the stable symbionts within the termite gut. Planctomycetes are rarely isolated from animals during cultivation attempts; with jellyfish^62^, sponges^63–65^ and - as described in this study - the termite gut as the only examples. In all cases, the interaction of the planctomycetal strains with their putative host remains enigmatic.
Comparison of plasmid sequences and identification of conserved plasmid genes
All characterized members of the family Isosphaeraceae have at least one and maximum five plasmids; however, the physiological role of maintenance of (several) extrachromosomal elements in the family remains enigmatic. The previously sequenced “K. aquadivae” EP7^T^ has four plasmids (with lengths 70, 58, 41 and 34 kbps), T. lichenicola P12^T^ has two (182 and 177 kbps) and strain TA3^T^ has four (81, 57, 52 and 11 kbps). Since core elements for plasmid replication and distribution in the family have not been identified, we used a “panplasmidome” approach to search for conserved features (Fig. 4B). No conserved genes were present in all plasmids and only two conserved plasmid-located genes were present in all three strains. These genes code for a putative nucleoside-diphosphate-sugar epimerase (WcaG) and a UDP-N-acetyl-D-mannosamine dehydrogenase (WecC). WcaG influences cell surface properties, virulence and extracellular enzyme production in the gamma-proteobacterium Pectobacterium carotovorum^66^. WecC is involved in the biosynthesis of enterobacterial common antigen, a surface polysaccharide specific for members of the family Enterobacteriaceae (class Gammaproteobacteria)^67^. Genes encoding putative ParA (ATPase) and ParB (Chromosome-partitioning protein) proteins of the ParABS system for plasmid partitioning were also identified. ParA-encoding genes were annotated on all plasmids except pTA3_4 (the smallest plasmid in the analysis) although in nine different gene clusters generated during the automated pangenome construction by anvi’o (indicating low sequence similarities). Putative ParB-encoding genes were only present on the two largest plasmids each in all three strains (i.e. absent in pEP7_3, pEP7_4, pTA3_3, pTA3_4) and fell in six clusters generated by anvi’o. PHASTEST did not yield any plasmid-located phage genes in either of the 10 analyzed plasmids. The results of the plasmidome analysis could be substantiated by clustering of the protein sequences of plasmid-encoded genes using CLANS (Clustering based on all-against-all BLAST + similarities). Apart from the above-mentioned proteins, the plasmids were rich in genes encoding putative serine/threonine and histidine protein kinases, tyrosine recombinases and proteins annotated as inositol dehydrogenases or inositol glycosyltransferases. As observed previously, plasmids in the family are heterogeneous^55^ and apart from the predicted ParA and ParB proteins that are likely involved in plasmid partitioning, the function of plasmid gene-encoded proteins remains to be elucidated.
Phenotypic characterization
On agar plates, strain TA3^T^ forms pink-pigmented, smooth and round colonies. When cultivated in larger volumes in limnic M3 medium in stirred bioreactors, the culture has a more orange to reddish color. With shaking, the strain did not form macroscopic aggregates and reached a final OD_600_ of 1.9-2.0 from a starting OD_600_ of 0.05. In limnic M3 medium, the strain grew over a temperature range of 18–28 °C and a pH range of 6.0–9.0. Under optimal conditions (pH 7.5, 24 °C), the strain reached a maximal growth rate of 0.026 h^− 1^, corresponding to a division time of ca. 27 h. The growth was slightly faster than that of T. lichenicola P12^T^ at 22°C (generation time 35 h)^22^. Strain TA3^T^ prefers slightly higher temperatures compared to its close relatives but all three strains failed to grow at temperatures > 28 °C (Table 1). The neutrophilic lifestyle of strain TA3^T^ is similar to that of “K. aquadivae” EP7^T^ but different from T. lichenicola P12^T^ that prefers slightly acidic conditions. In contrast to the strictly aerobic T. lichenicola P12^T^, strain TA3^T^ grew under oxygen-limited conditions. The strain could be passaged three times under anaerobic conditions in serum bottles, before turbidity did not increase anymore. Since the strain lacks genes encoding common enzymes involved in anaerobic respiration, e.g. nitrate, nitrite or sulfate reductase and formate dehydrogenase, it is likely that the observed growth under microaerobic conditions involves fermentation. The capability of the strain to grow under such conditions is in line with the finding that the hindgut of termites is typically a low-oxygen environment in which some oxygen can penetrate through diffusion from the termite tracheal system or via ingested materials.
Cells of strain TA3^T^ are spherical (Fig. 5A), occasionally oval and can form microscopic shapeless aggregates (Fig. 5B). Individual cells have a mean cell length and mean cell width of 2.5 ± 0.3 μm and 2.3 ± 0.2 μm (Fig. 5C), respectively. T. lichenicola P12^T^ and “K. aquadivae” EP7^T^ have a similar morphology, can also form aggregates, and have similar cell sizes^22,51^. Cells of strain TA3^T^ divide asymmetrically via “budding” where the daughter cell emerges as tiny round structure on one side of the cell, grows and eventually pinches off (Fig. 5); strains P12^T^ and EP7^T^ divide via the same mechanism. In other members of the phylum Planctomycetota the division can be further classified based on the localization/orientation of the division plane (polar or lateral)^4^. Such an orientation appears to be absent in spherical cells and the division mechanism/division plane placement mechanism cannot be categorized any further yet.
Fig. 5. Cell morphology and cell size of strain TA3^T^. (A) Microscopic images of cells of strain TA3^T^ captured with phase contrast (PhC) and differential interference contrast (DIC). (B) PhC and DIC microscopic image of cell aggregates as well as of individual cells. Dividing cells are indicated by an arrowhead pointing on daughter cells; scale bars are 2 μm. (C) Cell sizes determined in three replicates are indicated by three different colors; larger circles indicate the mean values of each replicate. The cell size is 2.5 ± 0.3 × 2.3 ± 0.2 μm (mean ± standard deviation), indicating spherical/oval cell shape.
Two additional typical traits of planctomycetes include invaginations of the cytoplasmic membrane giving room to an enlarged periplasmic space and a condensed nucleoid^3^. To investigate these characteristics in strain TA3^T^, cells were treated with Synaptored and DAPI, which stain membranes and DNA, respectively. Like for the model strain Planctopirus limnophila and “K. aquadivae” EP7^T^, cells of strain TA3^T^ displayed invaginations in the cytoplasmic membrane which occasionally spanned through the cytoplasm of the cells. Additionally, (multiple) intense DAPI spots in one cell could be observed pointing towards a condensed nucleoid as present in other planctomycetes (Fig. 6).
Fig. 6. Strain TA3^T^ displays invaginations of the cytoplasmic membrane and a condensed nucleoid. Individual cells and cells in aggregates display cytoplasmic membrane invaginations (stained with Synaptored, arrowheads) and therefore possess an enlarged periplasmic space. Cells possess a condensed nucleoid (stained with DAPI), occasionally cells display multiple smaller DAPI spots. PhC = Phase contrast, scale bars are 2 μm.
Conclusion
Strain TA3^T^ shows clear distinction from its closest relatives T. lichenicola P12^T^ and “K. aquadivae” EP7^T^ for both, genome-encoded/phylogenetic and phenotypic characteristics. The data supports a position for the novel strain within the genus Tundrisphaera but delineated from the sole species T. lichenicola. Hence, we propose a novel species, for which we introduce the name Tundrisphaera macrotermitis sp. nov.
Description of Tundrisphaera macrotermitis sp. nov.
Tundrisphaera macrotermitis (ma.cro.ter’mi.tis. N.L. gen. masc. n. macrotermitis, of the termite Macrotermes, from which the organism was isolated).
Aerobic chemoorganoheterotroph capable of growth under microaerobic conditions. Colonies are pink-pigmented. Cells are non-motile, spherical, and form shapeless aggregates or grow as single cells. The average cell size is 2.5 ± 0.3 μm and 2.3 ± 0.2 μm (length x width). Cells divide asymmetrically by budding. Optimum pH and temperature for growth are 7.5 (range 6.0–9.0) and 24 °C (range 18–28 °C), respectively. The type strain is TA3^T^ (= CECT 30560^T^ = STH00997^T^; the STH number refers to the Jena Microbial Resource Collection JMRC) and was isolated from the hindgut of a termite of the species Macrotermes natalensis sampled in Pretoria, South Africa. The type strain has a genome size of 7.23 Mbp (chromosome and four plasmids) and a DNA G + C content of 69.3%.
Emended description of Tundrisphaera lichenicola Kulichevskaya et al. 2017
Strain characteristics are as described before^22^ with the following modifications: The type strain has a genome size of 7.80 Mbp (chromosome and two plasmids) and a DNA G + C content of 64.7%.
Emended description of the genus Tundrisphaera
Characteristics of the genus are as previously described^22^ with the following modifications: Chemoheterotrophic aerobes, either strictly aerobic or capable of growth under microaerobic conditions. Neutrophilic or moderately acidophilic. High DNA G + C content from 65 to 70%.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wurzbacher, C. E. et al. Candidatus Uabimicrobium helgolandensis, a planctomycetal bacterium with phagocytosis-like prey cell engulfment, surface-dependent motility, and cell division. m Bio 15, e 02044-02024 (2024).10.1128/mbio.02044-24PMC 1148190639189742 · doi ↗ · pubmed ↗
- 2Zheng, R., Wang, C., Liu, R., Cai, R. & Sun, C. Physiological and metabolic insights into the first cultured anaerobic representative of deep-sea Planctomycetes bacteria. e Life 12, RP 89874 (2024).10.7554/e Life.89874 PMC 1094568838265071 · doi ↗ · pubmed ↗
- 3Klimek, D., Herold, M. & Calusinska, M. Comparative genomic analysis of Planctomycetota potential towards complex polysaccharide degradation identifies phylogenetically distinct groups of biotechnologically relevant microbes. bio Rxiv, 2024.2001.2010.575047 (2024).10.1186/s 12864-024-10413-z PMC 1113119938802741 · doi ↗ · pubmed ↗
- 4Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using Clustal W and Clustal X. Curr. Protoc. Bioinformatics, 2.3.1–2.3.22 (2003).10.1002/0471250953.bi 0203 s 0018792934 · doi ↗ · pubmed ↗
- 5Tarlachkov, S. & Starodumova, I. in Calculating the similarity value of the 16S r RNA gene sequences of prokaryotes or ITS regions of fungi. J Bioinform Genomics (2017).
- 6Rodriguez-R, L. M. & Konstantinidis, K. T. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J Preprints 4, e 1. (1900).
- 7Blin, K. et al. Anti SMASH 8.0: extended gene cluster detection capabilities and analyses of chemistry, enzymology, and regulation. Nucleic Acids Res gkaf 334 (2025).10.1093/nar/gkaf 334PMC 1223067640276974 · doi ↗ · pubmed ↗
- 8Kündgen, M. et al. Kueselia aquadivae gen. nov., sp. nov., the first member of the family Isosphaeraceae isolated from subsurface percolates. Sci. Rep. 15, 32243 (2025).10.1038/s 41598-025-17081-3PMC 1240250540890241 · doi ↗ · pubmed ↗