Loss of Bile Salt Export Pump (Bsep/Abcb11) Ameliorates Toxin-induced Hepatic Fibrosis via Suppression of Hepatocellular Jun Amino-terminal Kinase Signaling and Hepatic Stellate Cell Activation
Claudia D. Fuchs, Emmanuel D. Dixon, Philipp Königshofer, Thierry Claudel, Veronika Mlitz, Hubert Scharnagl, Tatjana Stojakovic, Thomas Reiberger, Michael Trauner

TL;DR
Removing a liver protein called Bsep reduces liver scarring in mice by blocking harmful cell signals and reducing inflammation.
Contribution
This study shows that Bsep loss prevents liver fibrosis by suppressing JNK signaling and HSC activation.
Findings
Bsep-/- mice showed reduced liver inflammation and fibrosis after toxin exposure compared to wild-type mice.
THBAs suppressed JNK activation in hepatocytes and reduced HSC activation markers in vitro.
Loss of Bsep altered bile acid profiles, which protected against toxin-induced liver damage.
Abstract
Loss of Bsep/Abcb11 results in a hydrophilic bile acid (BA) pool consisting of tetrahydroxylated BAs (THBAs) reducing cholestasis-induced liver injury. In this study, we investigated whether loss of Bsep may protect mice from development of toxin-induced liver fibrosis by directly impacting on hepatic stellate cell (HSC) activation. Wild-type (WT) and Bsep-/- mice were exposed to carbon tetrachloride (CCl4) or thioacetamide (TAA) for 4 weeks (3 injections per week) as models of toxin-induced liver fibrosis. In vitro, the human HSC line LX2 and immortalized human hepatocytes (IHHs) were challenged with TGFβ or 12S-HETE (arachonidonic acid derivate) with or without THBA treatment. Liver immunohistochemistry (IHC), immunofluorescence (IF), gene and protein expression, intrahepatic BA profile, luciferase activity, and 12S-HETE assays were performed. In contrast to WT mice, serum…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
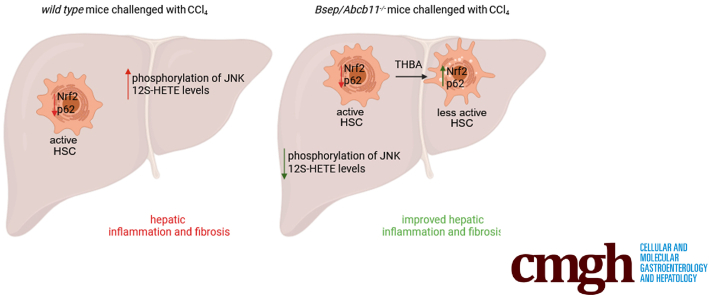 Figure 1
Figure 1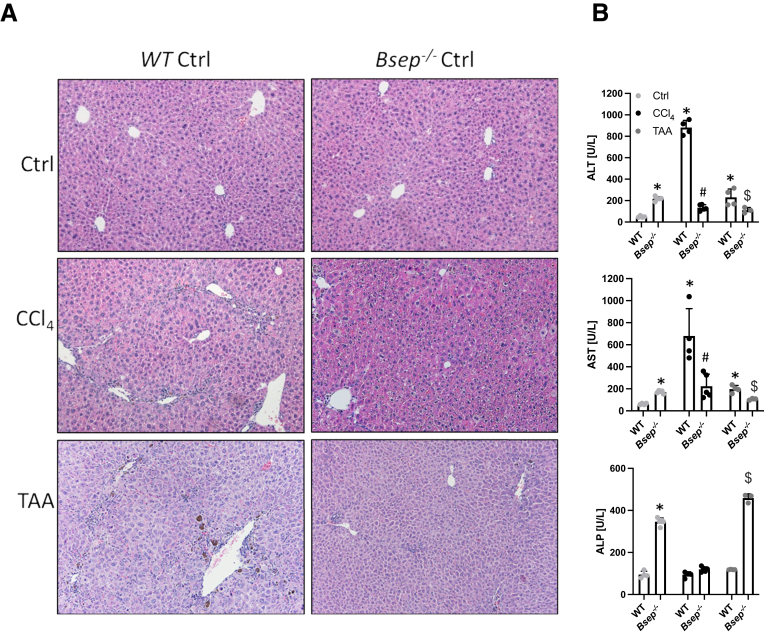 Figure 2
Figure 2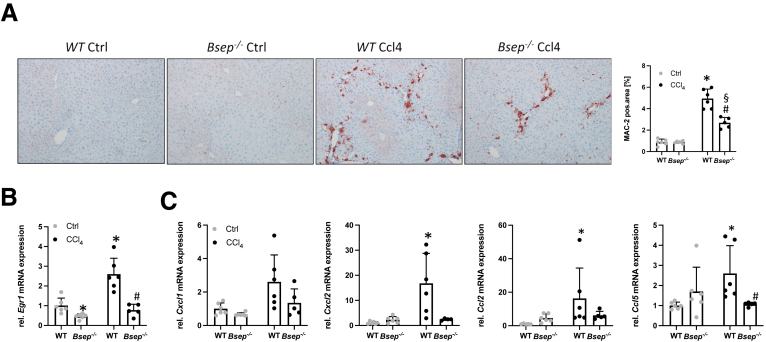 Figure 3
Figure 3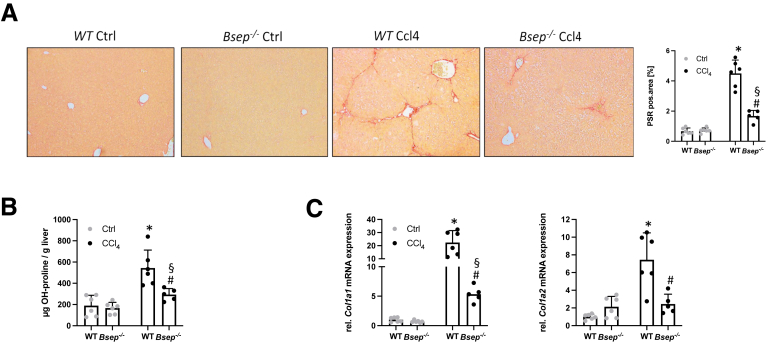 Figure 4
Figure 4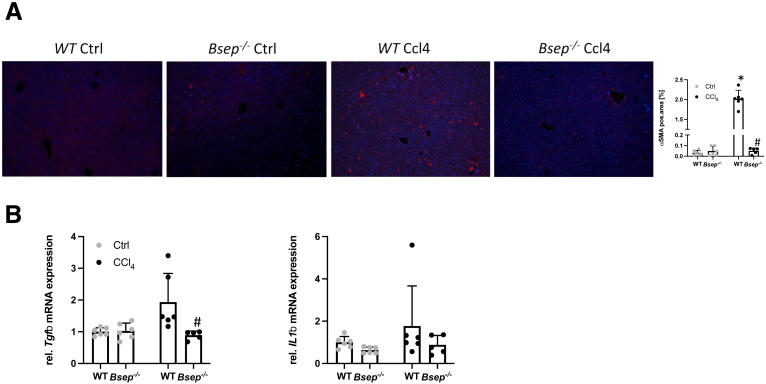 Figure 5
Figure 5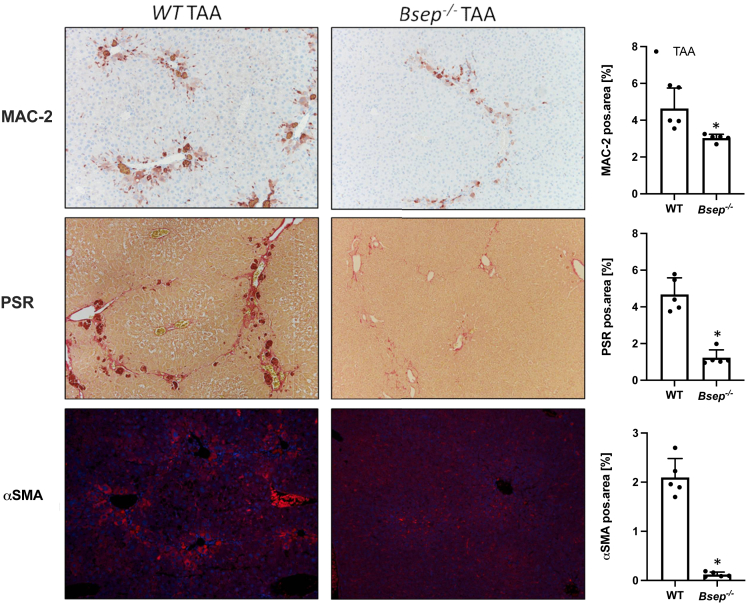 Figure 6
Figure 6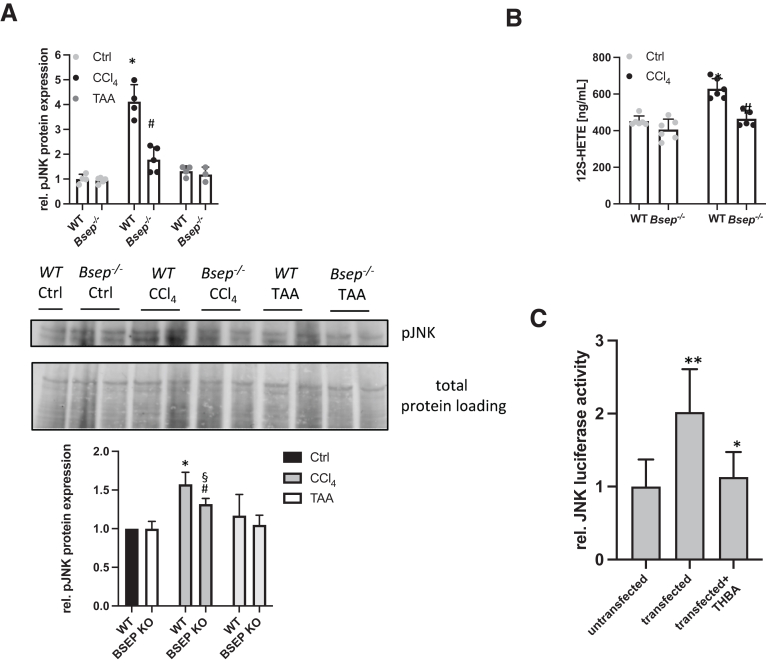 Figure 7
Figure 7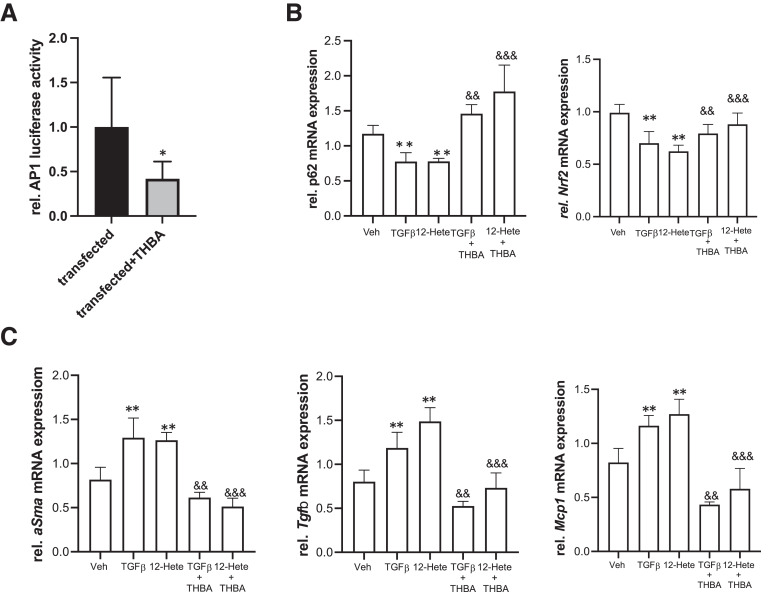 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPediatric Hepatobiliary Diseases and Treatments · Drug Transport and Resistance Mechanisms · Liver Diseases and Immunity
SummaryAbsence of Bsep/Abcb11, concomitant with the presence of tetrahydroxylated bile acids, protects mice from toxin induced hepatic fibrosis via suppression of hepatocellular jun amino-terminal kinase phosphorylation. Furthermore, tetrahydroxylated bile acids have a direct anti-fibrotic effect on hepatic stellate cells via restoring p62 and Nrf2 signaling.
Chronic unresolved liver injuries of different etiologies lead to an imbalance between production and dissolution of extracellular matrix (ECM). Hepatic stellate cells (HSCs) (shown to be the primary source of ECM) act in concert with hepatocytes and immune cells to aggravate scarring in response to liver injury.1 Apoptotic2^,^3 and stressed4 hepatocytes provoke inflammatory cell recruitment to the liver and the release of proinflammatory as well as pro-fibrotic cytokines such as transforming growth factor beta (TGFβ). Liver fibrosis may evolve further into cirrhosis and eventually hepatocellular carcinoma. To date, there is no specific therapy for patients suffering from hepatic fibrosis, and liver transplantation is often necessary when patients progress to cirrhosis portal hypertension and liver failure.5
Strategies that target bile acid (BA) signaling hold the potential for treatment of patients with liver disease.6, 7, 8 In addition to their anti-cholestatic effects,9 therapeutic BAs such ursodeoxycholic acid (UDCA) and its side chain shortened derivative norucholic acid (NCA, formerly norUDCA) were shown to be beneficial in thioacetamide (TAA)-induced fibrosis.10 Furthermore, NCA was shown to be protective against liver fibrosis provoked by Schistosoma mansoni infection.11 This observation was also recently extended to tetrahydroxylated bile acids (THBAs),12 which are very hydrophilic and thus potentially less toxic BA species formed in mice lacking bile salt export pump (Bsep/Abc11) and protecting these mice from (further) acquired cholestatic liver injury.13^,^14 Bsep^-/-^ animals have increased expression of Cyp3a11 and Cyp2b10,14 2 enzymes being key for BA hydroxylation.14 Hydroxylated BAs are secreted via alternative BA transporters Mrp3 and Mrp4 (both increased in Bsep^-/-^ animals14) into the blood and excreted via urine. Moreover, THBAs were shown to reduce cytokine secretion from hepatocytes due to suppression of early growth response 1 (Egr1), a critical driver of hepatic inflammation under cholestatic conditions. Thereby, THBA attenuated the numbers of infiltrating neutrophils and macrophages in livers of Mdr2^-/-^ mice as well as macrophage activation in vitro,13 indicating immunomodulatory effects of this BA, which may result in attenuation of hepatic fibrosis development.
In the present study, we aimed to investigate the hypothesis that THBAs may have a protective role in development of hepatic fibrosis by directly interfering with pathways involved in development of hepatic fibrosis and/or inhibiting activation of HSCs. Therefore, Bsep^-/-^ mice (with THBAs as one of the most prominent BA species in their BA pool13^,^14) were subjected to carbon tetrachloride (CCl_4_) and TAA treatment, commonly used to induce of toxin-mediated liver fibrosis.15
Results
Absence of Bsep Prevents Toxin-induced Liver Injury
After 4 weeks of CCl_4_ administration, Bsep^-/-^ mice did not show histological features of liver injury (Figure 1A), whereas in wild-type (WT) mice challenged with CCl_4_, pronounced histological features of hepatic inflammation and fibrosis were present (Figure 1A). Furthermore, serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) as markers of hepatocellular injury remained normal in CCl_4_-subjected Bsep^-/-^ mice but were profoundly elevated in CCl_4_-injected WT mice (Figure 1B). Although CCl_4_ in both WT and Bsep^-/-^ mice did not influence levels of alkaline phosphatase (ALP), serum BA concentration increased in WT mice after CCl_4_ application (Figure 1B). Of note, Bsep^-/-^ mice were also protected from TAA-induced liver injury reflected by improved liver transaminases (Figure 1A and B). However, liver injury was more severe in WT animals subjected to CCl_4_ than in the TAA setting (Figure 1B). Notably, intrahepatic BA profiling revealed that challenging Bsep^-/-^ mice with CCl_4_ led to a 40% reduction of THBA levels; TAA application even reduced intrahepatic THBA levels by 80% when compared with untreated Bsep^-/-^ mice (Table 1).Figure 1Loss of Bsep improves CCl_4_-induced liver injury. (A) Representative H&E images (×10 magnification). (B) Serum biochemistry reflects reduced levels of transaminases (ALT, AST) in Bsep^-/-^ mice subjected to CCl_4_ or TAA treatment compared with WT mice. ALP levels remained unchanged due to CCl_4_ challenge in WT mice and decreased in Bsep^-/-^ mice. TAA challenge led to increase of ALP levels in Bsep^-/-^ mice. ∗Significant difference from WT mice; ^#^significant difference from WT CCl_4_ mice; ^$^significant difference from WT TAA mice; P < .05.Table 1. Intrahepatic BA Profilingpmol/mgWT CtrlBsep^-/-^ CtrlWT CCl_4_Bsep^-/-^ CCl_4_WT TAABsep^-/-^ TAATCA11.72 ± 7.9018.92 ± 3.9124.72 ± 14.0721.06 ± 7.4422.91 ± 9.8130.38 ± 4.98b^,^cTUDCA0.17 ± 0.090.7 ± 0.140.26 ± 0.20.49 ± 0.110.39 ± 0.210.53 ± 0.07TCDCA0.29 ± 0.140.6 ± 0.130.55 ± 0.680.41 ± 0.100.46 ± 0.250.67 ± 0.14TDCA1.11 ± 0.320.05 ± 0.040.88 ± 0.390.04 ± 0.030.8 ± 0.440.09 ± 0.02ToMCA2.13 ± 0.8910.97 ± 3.17a2.93 ± 1.389.22 ± 3.925.43 ± 2.93a28.22 ± 4.41b^,^cTbMCA4.1 ± 1.9685.44 ± 19.54a14.76 ± 10.77115 ± 38.6611.31 ± 4.57a138.42 ± 27.13cCA0.45 ± 0.360.02 ± 0.010.85 ± 1.050.04 ± 0.011.12 ± 0.820.07 ± 0.05oMCA0.08 ± 0.040.17 ± 0.030.07 ± 0.040.21 ± 0.030.4 ± 0.280.58 ± 0.29bMCA0.03 ± 0.022.85 ± 0.63a0.31 ± 0.184.87 ± 1.211.04 ± 0.823.98 ± 1.83TTHBAn.d49.19 ± 17.880.7 ± 0.5320.92 ± 3.33a0.34 ± 0.2110.95 ± 4.76b^,^cTPHBAn.d0.44 ± 0.15n.d0.14 ± 0.03n.d0.06 ± 0.04Summ20.9 ± 11.4169.35 ± 23.76a46.03 ± 28.51172.46 ± 37.2944.71 ± 18.78214.16 ± 42.89b^,^cBA, bile acid; bMCA, betamuricholic acid; CA, cholic acid; CCl_4_, carbon tetrachloride; Ctrl, control; oMCA, omegamuricholic acid; TAA, thioacetamide; TbMCA, taurobetamuricholic acid; TCA, taurochenodeoxycholic acid; TCDCA, taurochenodeoxycholic acid; TDCA, taurodeoxycholic acid, ToMCA, tauroomegamuricholic acid; TPHBA, tauropentahydroxylated bile acid; TTHBA, taurotetrahydroxylated bile acid; TUDCA, tauroursodeoxycholic acid; WT, wild-type.aSignificant difference from WT mice; P < .05.bSignificant difference from Bsep^-/-^ mice; P < .05.cSignificant difference from Bsep^-/-^ CCl_4_ mice; P < .05.
Absence of Bsep Protects From CCl4-induced Liver Inflammation and Fibrosis
Next, we investigated markers of hepatic inflammation (Figure 2) and fibrosis (Figure 3) in more detail at the mRNA as well as protein level. Representative images of MAC-2/Galectin-3 immunohistochemistry (IHC) as well as computational analysis showed significantly less amounts of MAC-2 positive cells in liver of Bsep^-/-^ mice subjected to CCl_4_ in comparison to challenged WT animals (Figure 2A). mRNA expression of Egr1 (a key driver of hepatic inflammation, and known to be induced via CCl_4_16) was increased in WT CCl_4_-challenged mice and remained unchanged in Bsep^-/-^ mice upon CCl_4_ challenge (Figure 2B). Accordingly, gene expression profiling of Egr1 downstream targets such as Cxcl1, Cxcl2, Ccl2, and Ccl5 revealed a profound induction in WT mice upon CCl_4_ injection, whereas in CCl_4_-challenged Bsep^-/-^ mice, these markers remained at baseline levels, similar to unchallenged control mice (Figure 2C).Figure 2Loss of Bsep improves CCl_4_-induced liver inflammation. (A) Representative MAC-2/Galectin3 IHC images (×10 magnification) as well as computational analysis show reduced numbers of macrophages in the livers of Bsep^-/-^ mice. Real-time PCR was used to assess the mRNA expression of (B) Egr1 and its downstream targets (C) Cxcl1, Cxcl2, Ccl2, and Ccl5 as markers of inflammation. All of them were reduced in Bsep^-/-^ mice subjected to CCl_4_ treatment compared with WT mice. mRNA expression values were normalized against 36b4 levels and are shown relative to expression level in WT controls. ∗Significant difference from WT mice; ^#^significant difference from WT CCl_4_ mice; ^§^significant difference from Bsep^-/-^ mice; P < .05. Computational analysis of histological pictures was done via image J 1.51j8.Figure 3Loss of Bsep improves CCl_4_-induced liver fibrosis. (A) Representative PSR stainings (×10 magnification) as well as computational analysis show improved hepatic fibrosis in Bsep^-/-^ mice. (B) OH-proline content was reduced in Bsep^-/-^ mice subjected to CCl_4_ compared with challenged WT mice. (C) Real-time PCR was used to assess the mRNA expression of fibrotic marker collagen type I alpha 1 (Col1a1) and Col1a2, which were reduced in Bsep^-/-^ mice subjected to CCl_4_. mRNA expression values were normalized against 36b4 levels and are shown relative to expression level in WT controls. ∗Significant difference from WT mice; ^#^significant difference from WT CCl_4_ mice; ^§^significant difference from Bsep^-/-^ mice; P < .05. Computational analysis of histological pictures was done via image J 1.51j8.
Representative Picro-Sirius red (PSR) images as well as computational analysis displayed significantly lower collagen staining in Bsep^-/-^ animals after CCl_4_ application compared with challenged WT mice (Figure 3A). In addition, intrahepatic levels of hydroxyproline (OH-proline) were significantly lower in Bsep^-/-^ mice exposed to CCl_4_ in comparison with WT CCl_4_ animals (Figure 3B). In line, mRNA expression of fibrotic markers Col1a1 and Col1a2 was significantly lower in Bsep^-/-^ CCl_4_-injected mice compared with challenged WT mice (Figure 3C). In accordance with reduced hepatic fibrosis in Bsep^-/-^ CCl_4_-injected mice, numbers of alpha smooth muscle actin (αSMA)-positive cells (marker for activated HSCs), remained negligable when compared with WT CCl_4_-injected mice (Figure 4A). Subsequently, mRNA expression profiles of proinflammatory and profibrogenic cytokines such as Tgfβ and interleukin 1β (IL1β) known to be secreted from activated HSCs17^,^18 tended to be higher in WT mice upon CCl_4_ challenge (Figure 4B). Of note, Bsep^-/-^ animals challenged with TAA showed also reduced hepatic inflammation (MAC-2) and hepatic fibrosis (PSR) as well as reduced numbers of αSMA-positive cells when compared with WT TAA animals (Figure 5).Figure 4Loss of Bsep improves CCl_4_-induced activation of HSCs. (A) IF staining and computational analysis show reduced numbers of αSMA-positive cells in livers of Bsep^-/-^ mice subjected to CCl_4_ treatment compared with challenged WT mice. (B) Real-time PCR was used to assess the mRNA expression of markers of activated HSCs Tgfβ and IL1β, which were reduced in Bsep^-/-^ mice subjected to CCl_4_. mRNA expression values were normalized against 36b4 levels and are shown relative to expression level in WT controls. ∗Significant difference from WT mice; ^#^significant difference from WT CCl_4_ mice; P < .05. Computational analysis of histological pictures was done via image J 1.51j8.Figure 5Loss of Bsep improves TAA induced liver injury. Representative MAC-2/Galectin3 IHC images (×10 magnification) as well as computational analysis show reduced numbers of macrophages in the livers of Bsep^-/-^ mice. Representative PSR stainings (×10 magnification) as well as computational analysis show improved hepatic fibrosis in Bsep^-/-^ mice. IF staining and computational analysis show reduced numbers of αSMA-positive cells in livers of Bsep^-/-^ mice. ∗Significant difference from WT TAA mice; P < .05.
Lack of Bsep Attenuates CCl4-induced Phosphorylation of Jun Amino-terminal Kinases
Because CCl_4_ induces phosphorylation of jun amino-terminal kinases (JNK), leading to activation of the arachidonic acid signaling cascade, resulting in production of the proinflammatory trigger 12-hydroxyeicosatetraenoic acid (12S-HETE),19 we next investigated whether this key fibrogenic/pro-inflammatory mechanism is altered in Bsep^-/-^ mice. pJNK enzyme-linked immunosorbent assay (ELISA), and quantification of pJNK immune blot revealed that phosphorylation of JNK was induced upon CCl_4_ challenge in WT animals but not in Bsep^-/-^ mice (Figure 6A). Subsequently, serum concentrations of 12S-HETE were significantly lower in Bsep^-/-^ mice upon CCl_4_ injection when compared with challenged WT mice (Figure 6B). (TAA challenge did not interfere with levels of JNK phosphorylation, neither in WT nor in Bsep^-/-^ mice [Figure 6A]). Of note, in immortalized human hepatocytes (IHHs) transfected with the JNK-luciferase reporter, JNK activity was significantly reduced by treatment with THBA (Figure 6C), indicating that THBAs, known to be present in Bsep^-/-^ mice,13^,^14 may counteract the toxin-induced JNK signaling cascade, thus protecting from development of hepatic inflammation and fibrosis.Figure 6Loss of Bsep attenuates phosphorylation of JNK in vivo. Primary hepatocytes were isolated and (A) pJNK ELISA revealed increased phosphorylation of JNK in WT mice exposed to CCl_4_, whereas in Bsep^-/-^ mice pJNK levels remained unchanged. TAA exposure did not influence pJNK levels in WT and Bsep^-/-^ mice. Data are shown relative to WT Ctrl group. Western blot from primary hepatocytes revealed that loss of Bsep result in lower levels of JNK phosphorylation in the presence of CCl_4_. TAA exposure did not impact on pJNK levels. Western blot was quantified using ImageLab 10.0. ∗Significant difference from WT mice; ^#^significant difference from WT CCl_4_ mice; ^§^significant difference from Bsep^-/-^ Ctrl mice; P < .05. (B) 12 (S)-HETE ELISA showed reduced concentration of 12 (S)-HETE in serum of Bsep^-/-^ mice subjected to CCl_4_ compared with treated WT mice. ∗Significant difference from WT mice; ^#^significant difference from WT CCl_4_ mice; P < .05. (C) Human IHHs were transfected with a JNK-luciferase construct and treated with THBA. THBA attenuated JNK activation. ∗∗Significant difference from untransfected cells; ∗significant difference from transfected cells; P < .05.
THBA Counteracts HSC Activation In Vitro
Even though intrahepatic THBA levels were reduced due to CCl_4_ and TAA intoxication, in vitro, we investigated whether THBAs may have anti-fibrotic properties and thereby protecting Bsep^-/-^ mice from toxin-induced fibrosis. Therefore, LX2 cells were transfected with the AP1-luciferase reporter. AP1 activation, also contributing to activation of HSCs,20 was significantly reduced due to THBA treatment (Figure 7A).Figure 7THBA improves TGFβ and 12-HETE-induced activation of HSCs, in vitro. (A) The human HSC line LX2 was transfected with an AP1-luciferase construct and treated with THBA. THBA attenuated AP1activation. (B and C) LX2 cells were treated either with TGFβ or with 12S-HETE or with a combination of TGFβ and THBA or 12S-HETE and THBA. (B) Gene expression profiling showed that THBA restored p62 and Nrf2 expression, which was reduced due to TGFβ and 12S-HETE treatment. (C) mRNA expression of markers for activated HSCs, αSma, Tgfβ, and Mcp1 and were reduced in HSCs treated with TGFβ and THBA or 12S-HETE and THBA compared with mono-treatment of TGFβ and 12S-HETE. mRNA expression values were normalized against 36b4 levels and are shown relative to expression level in untreated controls. ∗∗Significant difference from control cells; ∗significant difference from CCl_4_-treated cells; ^&&^significant difference from TGFβ-treated cells; ^&&&^significant difference from 12S-HETE-treated cells; P < .05.
Furthermore, the LX2 cells were treated either with TGFβ or 12S-HETE (as a proinflammatory metabolite produced via the JNK signaling cascade) as potential activators of HSCs as well as with combinations either of TGFβ/THBA or 12S-HETE/THBA (Figure 7B and C). Although both TGFβ and 12S-HETE resulted in reduced expression of p62 and Nrf2 (known to have antioxidative as well as antifibrotic properties21), cotreatment with THBA restored expression levels of both markers (Figure 7B). Accordingly, THBA was able to significantly reduce TGFβ or 12S-HETE induced gene expression of profibrotic markers such as αSma, Tgfβ, and Mcp1 (Figure 7C). Together, these findings support our hypothesis that THBAs, as one of the predominant BAs in Bsep^-/-^ mice, protect them from toxin-induced liver injury by direct antifibrotic mechanisms.
Discussion
The present study demonstrates that mice lacking the canalicular BA export pump Bsep/Abcb11 are protected from toxin-induced liver fibrosis independent from potential anti-cholestatic actions. Mechanistically, we were able to show that pJNK signaling, known to be a key pathway in CCl_4_-induced liver injury,19^,^22 was repressed in Bsep^-/-^ mice with a hydrophilic BA pool. In line, THBA, a prominent BA species in Bsep^-/-^ mice,13^,^14 attenuated JNK signaling in human hepatocytes and proinflammatory/profibrogenic pathways in HSCs via restoring p62 and Nrf2 expression.
The hydrophobic BA taurocholic acid (TCA) was previously reported to directly activate phosphorylation of JNK.22 Because in Bsep^-/-^ mice the relative contribution of TCA to the total BA pool is rather negligible (14 and Table 1), this observation supports our finding of reduced hepatic JNK phosphorylation in Bsep^-/-^ mice. Given the predominance of THBA, a certain diluting effect attenuating proinflammatory effects of TCA (supposedly via JNK activation) may be involved in supporting hepatocecllular protection in Bsep^-/-^ mice. However, our in vitro findings in human hepatocytes also support a direct inhibitory effect of JNK phosphorylation via THBA as well.
Reduced phosphorylation/activation of JNK may also explain the low levels of 12S-HETE in serum of Bsep^-/-^ mice. Production of 12S-HETE, a member of the proinflammatory eicosanoids, secreted from immune cells as well as hepatocytes23 and responsible for recruitment of macrophages and neutrophils to the liver, is controlled by JNK activity.19 Our in vitro data support direct inhibitory effects of THBA on the pJNK/12S-HETE pathway.
Although EGR1 has been described as a stimulator of fibrogenesis,24^,^25 in the CCl_4_ model, it seems to play a hepatoprotective role during liver regeneration as a very early event in response to liver injury.16 In contrast to WT animals, in the Bsep^-/-^ mouse model Egr1 expression was not induced upon CCl_4_ challenge, which is in line with our previous findings that THBAs repress Egr1 expression.13 As such, in the Bsep^-/-^ CCl_4_ setting, Egr1 might not play a decisive role in counteracting development of hepatic fibrosis.
In the human HSC line LX2, THBA treatment reduced AP1-luciferase activity, restored p62 and Nrf2 expression, and ameliorated TGFβ and 12S-HETE-induced HSC activation, reflecting a possible direct anti-fibrotic effect of THBA. Lower levels of hepatic fibrosis found in CCl_4_- and TAA-injected Bsep^-/-^ mice might be a result of both direct antifibrotic and anti-inflammatory/immunomodulatory effects of THBA because proinflammatory cytokines and chemokines such as Tnfβ, IL1β, and Tgfβ secreted from activated macrophages, are known to activate HSCs.26
In conclusion, our results demonstrate that THBAs have direct anti-inflammatory as well as direct antifibrotic effects via counteracting the JNK signaling cascade and decreasing the production of cytotoxic metabolites such as 12S-HETE in hepatocytes. Moreover, they counteract HSC activation via restoring p62 and Nrf2 expression. Thus, future studies should evaluate if therapeutic strategies capable of increasing THBAs can ameliorate liver injury in patients with liver disease.
Materials and Methods
Animals
Male FVB/N WT and Bsep^-/-^ mice were kindly provided by the British Columbia Cancer Research Center27 and bred as reported previously.13^,^14^,^28^,^29 As described,30 age-matched 8-week-old male littermates were injected intraperitoneally either with CCl_4_ 1 mL/kg 25% solution in olive oil (Sigma-Aldrich) or 150 mg/kg TAA 3×/week for a time period of 4 weeks. From one batch of animals, primary hepatocytes were isolated after liver perfusion. For direct comparison, serum biochemistry, pJNK ELISA and pJNK Western blot were performed from this animal batch (n = 3–5). Histology, IHC/immunofluorescence (IF), RNA analysis, 12S-HETE assay and intrahepatic BA profile were performed from separate batches (n = 5–7). This animal study was approved by the Animal Ethics Committee of the Medical University of Vienna and the Federal Ministry of Science, Research, and Economy (BMWFW-66.009/008-WF/V/3b/2015) and was performed according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Serum Biochemistry and Histology
Serum biochemistry and histological staining (hematoxylin and eosin [H&E], PSR) was performed as described previously.31
IHC/IF
Detection of hepatic MAC-2 and αSMA was performed as described previously.32^,^33 In brief, IHC for MAC-2/Galectin-3 or IF for αSMA-positive cells was performed on formaldehyde (4% neutral-buffered)-fixed, paraffin-embedded liver sections using monoclonal mouse antibodies. Sections were deparaffinated, rehydrated, and digested with 0.1% protease. Endogenous peroxidase was blocked with 1% H_2_O_2_ in methanol. Specific binding of the MAC-2 antibody was detected using a biotinylated anti-rat IgG and the ABC-System with amino-9-ethyl-carbazole as substrate. For immunofluorescence, slides were incubated with a monoclonal anti-mouse α-SMA antibody (ab5694, Abcam). Slides were then washed and incubated with the secondary antibody: Alexa Fluor 594 goat anti rabbit IgG (Thermo Fisher Scientific).
Messenger RNA Analysis and Polymerase Chain Reaction
RNA isolation from liver, complementary DNA synthesis and real-time polymerase chain reaction (PCR) were performed as described previously.34 Oligonucleotide sequences are available upon request.
Luciferase Activity Assay in IHHs
IHH35 cells were seeded in a 24-well plate and transiently transfected with 0.3 μg/well of an JNK-luciferase construct (a gift from Dr C. Glineur, Pasteur Institute) using Fugene transfection reagent (Promega) in sterile Opti-modifed Eagle’s medium for 12 hours. Cells were incubated in complete media containing 10 μM THBA (3α, 6α, 7α, 12α tetrahydroxycholanoic acid; UHN Shanghai Research & Development) for 24 hours. Cells were lysed using a solution of 4% Triton X-100, glycylglycine 100 mM, MgSO4100 mM, and ethylene glycol tetra acetic acid 250 mM for 1 hour at room temperature on a shaker platform. The obtained extracts were then combined with the solution containing the substrate (luciferin 2.5 mM and adenosine triphosphate 20 mM; Sigma-Aldrich) and analyzed by a luminometer (Lumat LB950; EG&G Berthold).
Human HSC Culture
LX-2 cells, an immortalized HSC line,36 kindly provided by Prof S.L. Friedman (Mount Sinai School of Medicine) were cultured with Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies) supplemented with 5% non–heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin solution (EuroClone). Upon reaching approximately 80% confluency, LX-2 cells were detached from the culture flask using 0.25% trypsin-EDTA solution and reseeded in 6 well plates at a density of 1 × 10^6^ cells/well. Cells were cultured in DMEM supplemented with 4 ng/mL TGFβ (Sigma-Aldrich), 10 μM THBA (3α, 6α, 7α, 12α tetrahydroxycholanoic acid; UHN Shanghai Research & Development), and 100 nM 12S-HETE (Sigma-Aldrich) for 24 hours. For transfection, LX-2 were seeded in a 24-well-plate and transiently transfected with 150 ng/well of an AP-1-luciferase (from Takara Bio) construct using Fugene transfection reagent (Promega) in sterile DMEM without FBS for 12 hours. Complete medium was then added for 48 hours with/without 100 μM of THBA and the cells lysed (lysis solution: 4% Triton-X100, Glycyl-Glycine 100 mM, MgSO4 100 mM, EGTA 250 mM) for 30 minutes at room temperature on a shaker. The extracts were then analyzed with a luminometer (Lumat LB9507 EG&G Berthold) injecting 100 μL of a solution (Luciferin 2.5 mM and ATP 20 mM, Sigma-Aldrich).
Intrahepatic BA Analysis
Intrahepatic BA profiles were acquired using ultra-performance liquid chromatography tandem mass spectrometry as described previously.37
Phospho-JNK ELISA
Total protein was isolated from primary hepatocytes and concentration was obtained using Pierce BCA Protein Assay Kit from ThermoFisher Scientific. 100 μg protein were used to conduct the RayBio Phospho-JNK (Thr183/Tyr185) ELISA (Catalog Number PEL-JNK-T183) according to manufacturer’s protocol. Optical density (OD) was measured at 450 nm (OD, 450/100 μg protein).
Western Blotting
Total protein was isolated from primary hepatocytes, and concentration was obtained using Pierce BCA Protein Assay Kit from ThermoFisher Scientific. Target protein expression was normalized to total loaded protein amount, according to manufacturer’s instructions.
12S-HETE Assay
12S-HETE levels were measured in serum of animals according to the manufacturers’ manuals (Abcam; product number: ab133034).
Statistical Analysis
Results were evaluated using GraphPad Prism 8.4.1. Statistical analysis was performed using 2-way analysis of variance (ANOVA). Data were reported as means of 5 to 7 animals per group ± standard deviation (SD). A P value ≤ .05 was considered significant.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jiao J.Friedman S.L.Aloman C.Hepatic fibrosis Curr Opin Gastroenterol 2520092232291939696010.1097/mog.0b 013e 3283279668 PMC 2883289 · doi ↗ · pubmed ↗
- 2Bataller R.Brenner D.A.Liver fibrosis J Clin Invest 11520052092181569007410.1172/JCI 24282 PMC 546435 · doi ↗ · pubmed ↗
- 3Seki E.De Minicis S.Osterreicher C.H.TLR 4 enhances TGF-beta signaling and hepatic fibrosis Nat Med 132007132413321795209010.1038/nm 1663 · doi ↗ · pubmed ↗
- 4Allen K.Jaeschke H.Copple B.L.Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis Am J Pathol 17820111751862122405510.1016/j.ajpath.2010.11.026PMC 3070591 · doi ↗ · pubmed ↗
- 5Schnabl B.Scholten D.Brenner D.A.What is the potential role of antifibrotic agents for the treatment of liver disease?Nat Clin Pract Gastroenterol Hepatol 520084964971862873510.1038/ncpgasthep 1200 · doi ↗ · pubmed ↗
- 6Simbrunner B.Trauner M.Reiberger T.Review article: therapeutic aspects of bile acid signalling in the gut-liver axis Aliment Pharmacol Ther 542021124312623455586210.1111/apt.16602 PMC 9290708 · doi ↗ · pubmed ↗
- 7Trauner M.Fuchs C.D.Novel therapeutic targets for cholestatic and fatty liver disease Gut 7120221942093461572710.1136/gutjnl-2021-324305 PMC 8666813 · doi ↗ · pubmed ↗
- 8Trauner M.Fuchs C.D.Halilbasic E.Paumgartner G.New therapeutic concepts in bile acid transport and signaling for management of cholestasis Hepatology 652017139314042799798010.1002/hep.28991 · doi ↗ · pubmed ↗