Aldehyde or Hydrate? Investigation into the Oxidation of 5‐Formylcytosine Derivatives Using a Computational and Experimental Approach
Kuangjie Liu, Annika Menke, Fabian L. Zott, Domenic Mayer, Lena J. Daumann, Hendrik Zipse

TL;DR
This study explores how iron(IV)-oxido complexes oxidize 5-formyl nucleobases, revealing that hydrate forms play a key role in the reaction mechanism.
Contribution
The study identifies hydrate-mediated oxidation as a novel pathway for 5-formyl nucleobase oxidation by iron(IV)-oxido complexes.
Findings
Hydrate formation significantly influences the oxidation kinetics of 6-aza-derivatives.
Rate constants correlate with C—H bond dissociation values for 5-hydroxymethyl nucleobase oxidation.
Geminal diol intermediates accelerate hydrogen-atom transfer in the oxidation process.
Abstract
This study investigates the oxidation of 5‐hydroxymethyl and 5‐formyl nucleobases using an iron(IV)‐oxido complex that mimics the function of TET enzymes. A central question in this context is whether the oxidation of formyl substrates proceeds via the aldehyde or the hydrate form. To investigate the possible different reaction kinetics of these two forms, nucleobases containing a 6‐aza‐moiety are employed, giving rise to significantly more aldehyde hydrate as compared to the unaltered nucleobase. The concentration changes of substrates and products during oxidation were followed with 1H NMR spectroscopy. To analyze the kinetics of the oxidation reactions, a detailed numerical simulation of the stepwise sequential oxidation process is applied. 5‐Hydroxymethyl nucleobases are first oxidized to the respective 5‐formyl derivatives, which exist in equilibrium with their hydrate forms, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
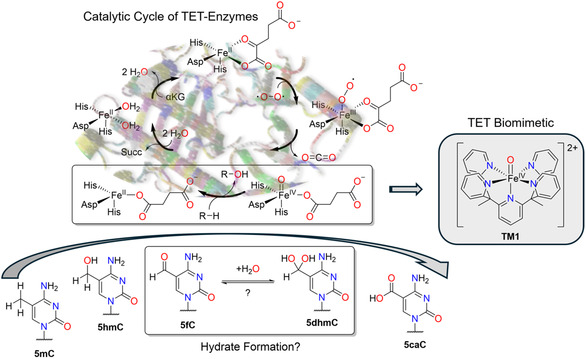 Figure 1
Figure 1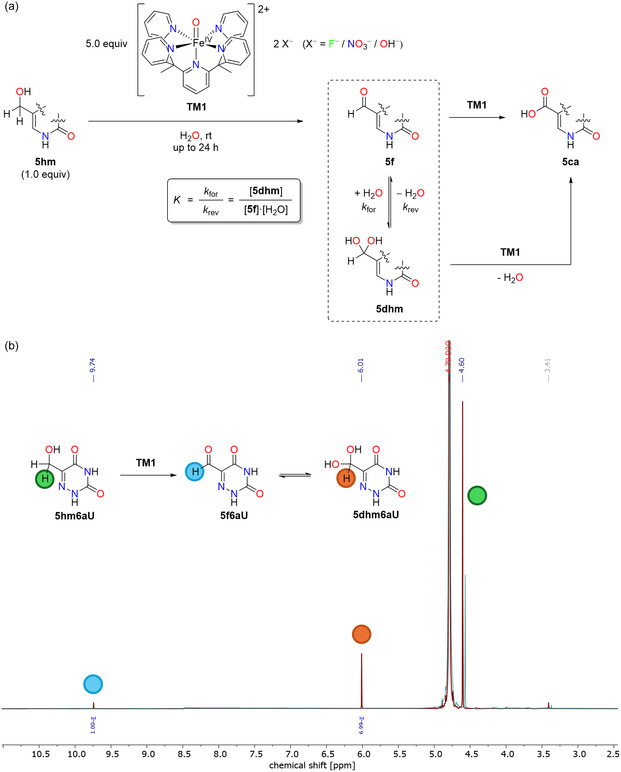 Figure 2
Figure 2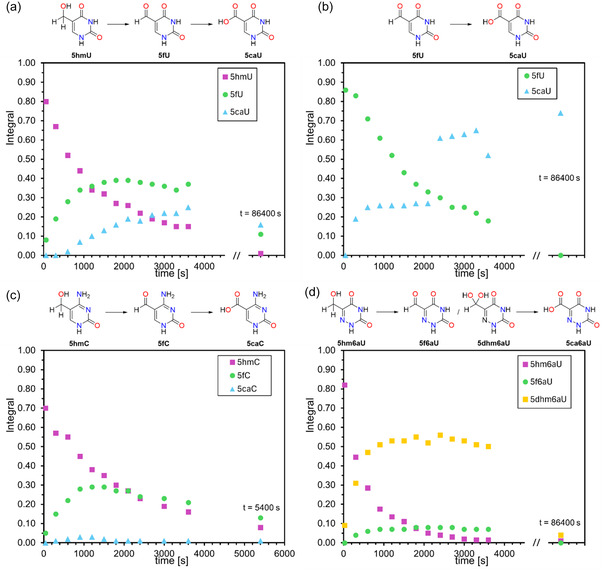 Figure 3
Figure 3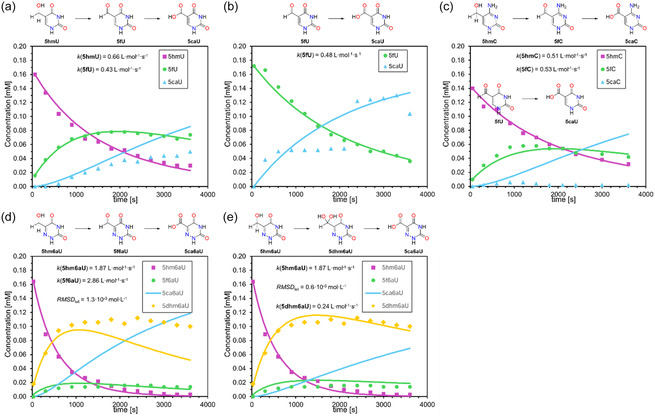 Figure 4
Figure 4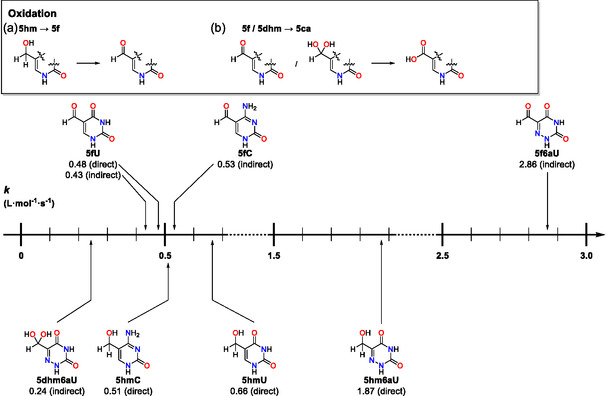 Figure 5
Figure 5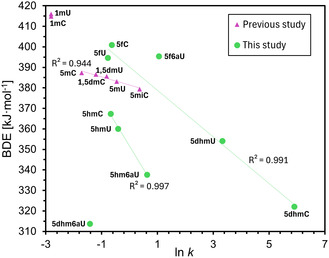 Figure 6
Figure 6| Nucleobase |
|
|
| |
|---|---|---|---|---|
| C–H BDE | 378.2 | 360.7 | 388.7 | – |
|
| 4.42 | 0.70 | 0.35 | |
| Base | Cascade | Step No. | Measurement |
|
|
|---|---|---|---|---|---|
| [L mol−1 s−1] | [L mol−1 s−1] | ||||
|
|
| 1 | direct | 0.66 | 0.02 |
|
| 2 | indirect | 0.43 | 0.01 | |
|
|
| 1 | direct | 0.48 | 0.01 |
|
|
| 1 | direct | 0.51 | 0.01 |
|
| 2 | indirect | 0.53 | 0.03 | |
|
|
| 1 | direct | 1.87 | 0.06 |
|
| 2 | indirect | 2.86 | 0.26 | |
|
| 2 | indirect | 0.24 | 0.01 |
| Base | BDE(C—H) |
| ln |
|---|---|---|---|
| [kJ mol−1] | [L mol−1 s−1] | ||
| Previous study[10] | – | – | – |
|
| 416.0 | 0.06 | −2.81 |
|
| 414.8 | 0.06 | −2.81 |
|
| 387.3 | 0.18 | −1.71 |
|
| 386.5 | 0.30 | −1.20 |
|
| 385.7 | 0.44 | −0.82 |
|
| 383.1 | 0.63 | −0.46 |
|
| 379.4 | 1.44 | +0.36 |
| This study | – | – | – |
|
| 401.0 | 0.53 | −0.64 |
|
| 395.4 | 2.86 | +1.05 |
|
| 394.7 | 0.46 | −0.78 |
|
| 367.4 | 0.51 | −0.67 |
|
| 360.0 | 0.66 | −0.42 |
|
| 354.1 | (27.66) | (+3.32) |
|
| 337.7 | 1.87 | +0.63 |
|
| 322.1 | (364.23) | (+5.90) |
|
| 313.8 | 0.24 | −1.43 |
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpigenetics and DNA Methylation · RNA modifications and cancer · Adenosine and Purinergic Signaling
Introduction
1
Epigenetics expands the scope of biological information storage beyond the sequence of canonical DNA bases.^[^ 1 ^]^ The most prominent epigenetic alteration is the methylation of cytosine (C) to 5‐methylcytosine (5mC).^[^ 2 ^]^ Methylated CpG sites act in the regulation of gene expression by influencing repressor protein^[^ 3 ^]^ and transcription factor binding,^[^ 4 ^]^ as well as chromatin dynamics.^[^ 5 ^]^ The reversibility of DNA modifications enable changes during an organism's development and its adaptation to environmental circumstances.
To this end, demethylation is largely orchestrated by the α‐ketoglutarate‐ and O_2_‐dependent nonheme ten‐eleven translocation (TET) dioxygenases.^[^ 6 ^]^ TET enzymes can sequentially oxidize 5mC to their respective hydroxymethyl (5hmC), formyl (5fC), and carboxylic acid (5caC) derivatives. These moieties are prone to passive demethylation during replication, active demethylation via the base excision repair (BER) pathway, or the less established direct deformylation and decarboxylation.^[^ 7 ^]^ In the generally accepted oxidation mechanism of TET enzymes,^[^ 8 ^]^ the hydrogen atom transfer (HAT) step is viewed as rate‐limiting, but not all steps are fully elucidated. In particular, a potential hydrate formation of 5fC toward 5‐dihydroxymethylcytosine (5dhmC) is discussed concerning further TET‐dependent oxidation and specific oligonucleotide binding events (Figure 1).^[^ 9 ^]^ The potential involvement of HAT processes is supported by theoretically calculated C—H bond dissociation energies (BDEs) and their qualitative correlation with HAT efficiencies.^[^ 10 ^]^ However, due to conformational restraints, this is not observed for substrates in the catalytic pocket of TET enzymes, as illustrated by empirical data from Xu et al.^[^ 11 ^]^ and calculations by Luo et al. (Table 1).^[^ 12 ^]^
*Catalytic cycle of TET enzymes. Highlighted hydrogen atom transfer step (HAT) and sequential array of oxidized substrates from 5mC to 5caC. Structure of TET biomimetic (TM1) used as model complex.[
20
]*
The simplified TET biomimetic [Fe^IV^(O)(Py_5_Me_2_H)]^2+^ (TM1), first published by Chang et al.^[^ 13 ^]^ was shown to oxidize 5mC as nucleobase,^[^ 14 ^]^ as nucleoside or as oligomeric nucleotide.^[^ 15 ^]^ Here, the calculated BDE values shown in Table 1 correspond well with the found reaction rates of 5mC and 5hmC. Accumulation of 5fC during the oxidation was observed, but no validated reaction rate for the substrate has yet been reported (Table 1).^[^ 14 ^]^ As the 5dhmC geminal diol was detected in (+)‐MS experiments^[^ 9 ^b]^ and has been implicated as an explanation of different base‐flipping kinetics,^[^ 16 ^]^ a transient hydrate formation during TET‐mediated 5fC oxidation seems possible. 5dhmC forms in a pH‐dependent manner and was found at 0.5% under acidic conditions.^[^ 9 ^]^ In contrast, the hydrate form of the noncanonical nucleotide 5‐formyl‐6‐aza‐cytidine (1rb5f6aC) was reported to be comparatively abundant at 20% by Carell et al.^[^ 17 ^]^ Kinetic measurements on selected substrates alongside the respective BDE value calculations might therefore aid in evaluating the importance of the hydrate adduct for 5fC oxidation with potential implications regarding the relevance of direct deformylation and decarboxylation of methylcytosine derivatives.
Results and Discussion
2
Analytical Data
2.1
Following the procedure illustrated in Figure 2, each oxidation reaction employed a substrate concentration of 0.2 mM and a fivefold excess (1.0 mM) of iron‐complex TM1 at 25 °C. Synthesis of the iron complex followed literature procedures.^[^ 13 ^,^ 18 ^]^ After anion exchange to remove excess cerium nitrate species, TM1 is obtained as an aqueous solution (10 mM) that was always used in situ and assumed to exist as a mixed anion system (including fluoride, nitrate, and hydroxide ions). Reaction kinetics were measured by taking samples at selected reaction times, followed by a work‐up step employing adsorptive filtration through a short silica gel column, and ^1^H NMR analysis of the filtrate using pyrazine as internal standard. The four species identified spectroscopically in oxidation experiments starting with 5hm nucleobases include the starting material itself, the 5f derivative in equilibrium with its hydrate form 5dhm, and 5ca as the endpoint of the oxidation cascade as shown in Figure 2a. The time‐dependent integrals observed for oxidation reactions starting from 5hmU, 5fU, 5hmC, and 5‐hydroxymethyl‐6‐aza‐uracil (5hm6aU) have been collected in Figure 3. The sum of reactant and product integrals decreases during the reaction, most evidently at longer reaction times. We attribute this to the loss of nucleobases on the silica gel column, minor side reactions, and partial degradation caused by the reactive iron(IV)‐oxido species. While no additional signals were identified in the ^1^H NMR spectra, it is likely that proton‐deficient or paramagnetic products are formed in trace amounts. Combination of the integrated signal intensities with the known internal standard concentrations provides time‐dependent concentrations of allreactants/intermediates/products and thus the basis for the subsequent kinetic analysis. Inspection of the results obtained for the oxidation of 5hmU (Figure 3a) indicates 5fU as the only detected intermediate and 5caU as the final product. Effectively, the same result is obtained when starting the oxidation from 5fU as shown in Figure 3b. While no hydrate could be detected as a transient intermediate in the oxidation of 5hmC (Figure 3c), this species is the most abundant intermediate detected in the oxidation of 5hm6aU (Figure 3d). Importantly, 5fC and 5fU both have extremely small hydration equilibria (*K *< < 10^−3^),^[^ 19 ^]^ which explains why their hydrate forms were not observed under the reaction conditions used in this study. By contrast, a hydrate content of 20% has been reported for 5‐formyl‐6‐aza‐cytidine (1rb5f6aC), which corresponds to *K *= 4.5 × 10^−3 ^L mol^−1^.^[^ 17 ^]^ Since uracil derivatives typically hydrate about 20‐fold more readily than their cytosine analogs, we extrapolated K(5f6aU) ≈ 9.0 × 10^−2 ^L mol^−1^, which predicts an equilibrium hydrate content of 83% (see Figure S1 in Supporting Information). Strikingly, our NMR experiments on 5f6aU oxidation show a constant diol content of ≈88%, in excellent agreement with the estimated 83% hydrate content (Figure 3d). The fact that this ratio remains essentially unchanged over the entire reaction time, where 5f6aU is being continually oxidized by TM1 to the final 5ca6aU product, demonstrates that the aldehyde‐hydrate equilibrium is established much faster than the oxidation itself. This justifies treating the hydration step as a rapid pre‐equilibrium in our kinetic simulations (see below) and highlights the mechanistic role of the geminal diol in directing the subsequent iron(IV)‐oxido mediated oxidation of 5f6aU.
a) Reaction mechanism for the oxidation from 5‐hydroxymethyl (5hm) via 5‐formyl (5f) and 5‐dihydroxy (5dhm) to 5‐carboxyl (5ca) with biomimetic iron(IV)oxido complex TM1 in water at room temperature. The equilibrium constant between 5f and 5dhmU is formulated as K = k for/k rev. b) 1H NMR spectrum in D2O of 5hm6aU oxidation. Integration of the aldehyde and hydrate peaks revealed a distribution 5f6aU:5dhm6aU = 1:7 (88% hydrate). Reaction conditions: [5hm6aU] = [TM1] = 5 mM, H2O, 25 °C, 1 h.
Raw 1H NMR integrals as observed for the oxidation cascade of a) 5hmU, b) 5fU, c) 5hmC, and d) 5hm6aU.
Kinetic Modeling
2.2
Numerical microkinetics simulations with the COPASI package where employed to fit the time dependent concentrations of all species to the general oxidation mechanism shown in Figure 2a. The reaction steps considered in the kinetic models also include the slow first‐order self‐deactivation of the TM1 oxidant to its reduced form (TM2) quantified in independent experiments ([TM1]_0 _= 1.0 mM, k _TM _= 1.75·10^−5 ^s^−1^). Using the oxidation cascade of 5hmU via 5fU to 5caU as an example, thus yields the following kinetic model
step 0:
step 1:
step 2:
Preliminary experiments indicated that ommitting the last measurement at very long reaction times (*t *= 86,400 s) and treating the initial substrate concentration as a variable (rather than a fixed quantity) gave the best results for all four substrates. This is not surprising as some susbtrates reacted faster than others and within the used setup, starting concentrations at the first measurement point are thus slightly different. Initial fitting of the rate constant for 5hmU oxidation to the decay of this species, and subsequent use of the optimized value in fitting the rate constant for 5fU oxidation to the concentrations of all species provided the most robust approach for evaluation of the overal kinetic scheme and gave k(5hmU) = 0.66 ± 0.02 L mol^−1 ^s^−1^ and k(5fU) = 0.43 ± 0.01 L mol^−1 ^s^−1^.
Data Analysis
2.3
The rate constants derived from the primary data shown in Figure 3 for the oxidation of 5hmU, 5hmC, and 5hm6aC are collected in Table 2. The measurement type “direct” indicates that the rate constant was derived as the first step of the overal oxdiation cascade, while “indirect” indicates a rate constant for oxidation of a transient intermediate.
In how far the direct and indirect measurement types yield different values was tested for the oxidation of 5fU, whose direct measurement gave k(5fU) = 0.43 ± 0.01 L mol^−1 ^s^−1^. Determination of the same rate constant as the second step of the oxdiation cascade starting from 5hmU gave a closely similar value of k(5fU) = 0.48 ± 0.01 L mol^−1 ^s^−1^, which demonstates the robustness of our kinetic analysis. The fidelity of the rate constant determinations can also be inferred from the standard deviations collected in Table 2, and also from visual inspection of Figure 4, where experimentally measured turnover data is combined with the simulated reaction progress as defined by the rate constants. The predicted decay of 5hmU, as well as the intermediate rise and subsequent decrease of 5fU as a transient intermediate, are quite well reproduced by the simulated turnover cuves. In contrast, the accurate quantification of 5caU suffers from the chosen work‐up method, as is reflected in the kinetic fits and raw data shown in Figure 4a,b. This implies that accurate kinetic data will only be obtained by following the concentrations of 5hmU and 5fU, but not that of 5caU. The same observation can be made for 5hmC as the initial substrate, where rate constants for the oxidation of 5hmC and 5fC are close to those obtained for the uracil derivatives before, and where quantification of the final 5caC product is again not possible in a quantitative manner. Kinetic analysis of 5hm6aU oxidation differs from the first two substrates in that the first step of the oxidation cascade is much faster than for the non‐aza parent systems with k(5fU) = 1.87 ± 0.06 L mol^−1 ^s^−1^, and where 5f6aU and 5dhm6aU are both detected as true intermediates of the oxidation cascade.
Overlay of simulated data with original data for the oxidation cascade of a) 5hmU, b) 5fU, c) 5hmC, d) 5hm6aU via 5f6aU, and e) 5hm6aU via 5dhm6aU with the determined rate constants and the overall RMSDtot value for all observed species.
The experimentally determined second order rate constants k for the oxidation step converting 5hm‐ to 5f‐, and 5f‐ to 5ca‐nucleobases are summarized in a graphical manner in Figure 5. Oxidation of 5hmC and 5hmU proceeds at largely similar reaction rates of 0.51 and 0.66 L mol^−1 ^s^−1^, respectively. Oxidation of 5hm6aU to its 5f‐derivative is significantly faster with a rate constant of 1.87 L mol^−1^ s^−1^, indicating that substitution at the C6 position with nitrogen (6‐aza‐substitution) accelerates the transformation significantly.
Second‐order rate constants (L mol−1 s−1) for the single‐step oxidation of a) hydroxymethyl‐ and b) formyl‐substituted nucleobases by iron complex TM1.
Kinetic analysis of the oxidation of 5f nucleobases is complicated by the possible involvement of aldehyde hydrates in the oxidation cascade, even when direct detection of these hydrates by ^1^H NMR measurements is not possible or inconclusive. This is the case for 5fU, where the known equilibrium constant for hydrate formation amounts to K(5fU) = 2.94·10^−4 ^L mol^−1^.^[^ 19 ^]^ Kinetic analysis of the oxidation of 5fU thus assumes no involvement of the respective hydrate form and yields rate constants of 0.48 L mol^−1 ^s^−1^ (direct method) and 0.43 L mol^−1 ^s^−1^ (indirect method), both of which are identical within experimental error and only marginally lower than that for 5hmU at 0.66 L mol^−1 ^s^−1^ (Table 2). For 5fC, the hydration equilibrium is even less favorable than for 5fU. Equilibrium constants have been proposed based on the limit of detection (*K *= 4.05 ·10^−5^) and the limit of quantification (*K *= 1.22 × 10^−5^) in ^1^H NMR measurements, yielding an average value of *K *= 2.64 × 10^−5 ^L mol^−1^.^[^ 19 ^]^ Assuming that oxidation of 5fC proceeds without the involvement of its hydrate form yields k(5fC) = 0.53 L mol^−1 ^s^−1^, which is only marginally faster than oxidation of 5fU.
The oxidation of 5f6aU is mechanistically more complex in that two plausible reaction pathways exist for its conversion to 5ca6aU. The first involves oxidation of 5f6aU without the involvement of any 5dhm6aU oxidation. This yields a rate constant of k(5f6aU) = 2.86 L mol^−1 ^s^−1^ together with a comparatively large cumulative root mean square deviation (RMSD_tot_) value (Figure 4). The second pathway assumes that only 5dhm6aU is oxidized out of the hydration equilibrium with 5f6aU, which yields k(5dhm6aU) = 0.24 L mol^−1 ^s^−1^ together with a more favorable RMSD_tot_ value (Figure 4). This rate constant is counterintuitively small, but results from the comparatively high concentration of 5dhm6aU in the hydration equilibrium. This further strengthens our assumption that the oxidation over the hydrate route better describes the oxidation cascade. Kinetics simulations with both oxidation pathways active simultaneously met with significant numerical problems but confirm that the aldehyde‐hydrate equilibrium is established on a timescale much faster than oxidation, and that most of the substrate is oxidized via the hydrate form. Consequently, it is both chemically and kinetically more appropriate to treat hydration as a rapid pre‐equilibrium and to describe the system by a single, effective oxidation rate of the hydrated species. This insight underpins our final kinetic model and reinforces the mechanistic conclusion that geminal‐diol formation is an obligate and dominant feature of 5f6aU oxidation by the iron(IV)‐oxido biomimetic complex TM1. The results also underscore how small changes in nucleobase structure can markedly alter the oxidation kinetics. Electron‐withdrawing substituents modulate the susceptibility of the 5hm group to undergo oxidation, and 5hm6aU is the most rapidly oxidized to its 5f form among the nucleobases tested, whereas 5hmC and 5hmU proceed at more moderate but still comparable rates.
Mechanistic Insights from Comparison to BDE(C—H) Values
2.4
How C—H bond abstraction from formyl hydrates relates to that from the parent aldehydes can be assessed by inspection of the respective BDE(C—H) values. Using the same theoretical approach as employed in a recent reactivity analysis of 5‐methyl substituted nucleobases,^[^ 10 ^]^ we have now computed the BDE(C—H) values for the 5hm, 5f, and 5dhm derivatives of the pyrimidine bases studied here (Table 3). For each of the bases studied here, we find the BDE(C—H) values to be largest for the formyl C—H bonds, followed by those for the hydroxymethyl C—H bonds, and lowest for the C—H bonds in the aldehyde hydrates. Closer inspection of Table 3 shows that the BDE(C—H) values for the three formyl group C—H bonds in 5fC, 5f6aU, and 5fU are closely similar (within 6 kJ mol^−1^), while this is not so for the hydrates of these nucleobases. For these latter systems, the weakest C—H bond is found for 5dhm6aU at BDE(C—H) = 313.8 kJ mol^−1^, which is more than 40 kJ mol^−1^ lower than that for 5dhmU at BDE(C—H) = 354.1 kJ mol^−1^. Despite this apparent lack of systematic bond energy trends, we can nevertheless employ the computed BDE(C—H) values for correlation analysis with the experimentally determined oxidation rate constants. These are also collected in Table 3 together with their logarithmic forms. For 5fU oxidation, an average rate constant k(5fU) = 0.46 L mol^−1 ^s^−1^ was used. For the other species, there is only one value each.
As already mentioned above, the equilibrium constants for the hydration of 5fC and 5fU are extremely low, but we can nevertheless explore the scenario where the oxidation of these systems proceeds solely through the respective hydrate forms. For 5fC the average value for the equilibrium constant K(5fC) = 2.64·10^−5 ^L mol^−1^ was used, while for 5fU we employ the known equilibrium constant K(5fU) = 2.94·10^−4 ^L mol^−1^.^[^ 19 ^]^ For 5f6aU the equilibrium constant has not been determined under stationary conditions, and we therefore employ here the extrapolated value of K(5f6aU) = 9.0 × 10^−2 ^L mol^−1^ mentioned already above. In dilute solutions, it is practical to consider that the equilibrium between the formyl and hydrate is rather quickly established, which can be reflected in the kinetics simulations by assuming a fast forward rate constant of k _for _= 1000 L mol^−1 ^s^−1^ in combination with a reverse rate constant k rev such that the ratio of both yields the equilibrium constant. The overall kinetics scheme then is
step 0:
step 1:
step 2:
step 3:
Applying this kinetic model yields rate constants for 5dhmU of *k *= 26.2 ± 0.4 L mol^−1 ^s^−1^ for the indirect method and *k *= 29.2 ± 0.7 L mol^−1 ^s^−1^ for the direct method, the average of which is k(5dhmU) = 27.7 L mol^−1 ^s^−1^, while for 5dhmC we obtain a notably large value of k(5dhmC) = 364.2 ± 20.7 L mol^−1 ^s^−1^ (Table 3). The rather large rate constants for 5dhmU and 5dhmC simply result from the rather low concentration of the respective hydrates under equilibrating conditions. Assuming that reaction proceeds through the respective hydrates does not lead to an improved fit of the turnover curves to the kinetic model for these two species. This is different for oxidation of 5f6aU where the oxidation route via the hydrate form 5dhm6aU provides the best fit for the turnover curves of 5f6aU and 5dhm6aU.
With the newly derived rate constants for the hydrate forms in hand, we can explore a possible correlation with the respective BDE(C—H) values (Figure 6). For the correlation of the calculated BDE values with the natural logarithm of the experimentally measured second‐order rate constants k, we observe a linear relationship for the three 5hm species (R ^2 ^= 0.997). As expected, the correlation predicts an increase in oxidation rate with decreasing BDE(C—H) values. Extension of this correlation to significantly higher BDE(C—H) values appears to cross through data points for the oxidation of methylated cytosine and uracil derivatives determined in earlier studies,^[^ 10 ^]^ but not those for the three formyl derivatives investigated in the current study. If we were to include 5fC and 5fU in the data set, the R ^2^ value drops to 0.677, suggesting a less satisfactory correlation. Despite the structural diversity, a second inverse correlation between BDE and ln k (R ^2 ^= 0.991) appears to include 5fC and 5fU with their hydrated counterparts 5dhmC and 5dhmU, but not the respective 6‐aza variants 5f6aU and 5dhm6aU. This may again suggest that the oxidation of the aza‐substituted 5f6aU proceeds through a different mechanism. We may thus conclude that oxidation proceeds through the hydrate for the species that exhibit high hydrate‐formation, combined with the fact that the BDE for the C—H of the geminal diol is much lower than the original formyl group, thereby facilitating the oxidation.
*Correlation of the experimentally derived rate constants for the oxidation cascades with the calculated bond dissociation energies (BDEs) determined in this study in comparison with our previous study.[
10
]*
Conclusion
3
We have combined quantitative NMR experiments, numerical kinetic modeling, and density functional theory calculations to clarify the mechanism of side‐chain oxidation in 5‐substituted nucleobases by a biomimetic iron(IV)‐oxido complex (TM1). Time‐resolved ^1^H NMR measurements allowed monitoring the stepwise conversion of 5‐hydroxymethyl (5hm) to 5‐formyl (5f) and ultimately to 5‐carboxyl (5ca) derivatives for a series of substrates (5hmU, 5fU, 5hmC, and 5hm6aU). By accounting for the oxidant's slow self‐deactivation and properly selecting initial and final turnover points, we obtained robust second‐order rate constants k spanning two orders of magnitude from 5hmC to 5hm6aU, 0.51–1.87 L mol^−1 ^s^−1^, for the direct 5hm to 5f step. Notably, the 6‐aza substitution in 5hm6aU accelerates hydrogen‐atom abstraction by almost threefold, underscoring the influence of heterocycle electronics on reactivity. For the oxidation from 5f to 5ca, we distinguished two scenarios: the direct oxidation of 5f or the oxidation of the hydrate counterparts (indirect pathway). We found that the numerical description of direct oxidation for the nucleobases with low hydrate formation tendency, 5fC and 5fU, is sufficient. However, for the nucleobase with significant hydration, 5f6aU, the oxidation via 5dhm6aU better fits the whole oxidation cascade. Complementary DLPNO‐CCSD(T)/CBS calculations of C—H BDEs reveal an excellent inverse correlation (R ^2 ^≈ 0.99) between bond strength and oxidation rate across the 5hm series, supporting that hydrogen‐atom abstraction is rate‐determining. For formyl substrates (5fU and 5fC) with sufficiently low hydration equilibria, a linear correlation with their hydrates (5dhmU and 5dhmC) may be understood such that minimal hydration doesn’t change the reaction mechanism. However, the significantly increased level of hydration of 5f6aU suggests that the rate‐determining step from C—H abstraction at the aldehyde switches to that at its hydrate counterpart 5dhm6aU. Together, this comprehensive study shows that direct H‐atom abstraction governs 5hm oxidation, while hydrate‐mediated pathways can dominate 5f oxidation when the aldehyde hydration is thermodynamically favorable. This mechanistic duality not only has important implications for TET‐catalyzed demethylation in epigenetics but also provides design principles for tailoring biomimetic oxidants toward selective C—H activation in nucleic acids.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1E. R. Gibney , C. M. Nolan , Heredity 2010, 105, 4.20461105 10.1038/hdy.2010.54 · doi ↗ · pubmed ↗
- 2a) L. D. Moore , T. Le , G. Fan , Neuropsychopharmacology 2013, 38, 23;22781841 10.1038/npp.2012.112PMC 3521964 · doi ↗ · pubmed ↗
- 3E. Ballestar , A. P. Wolffe , Eur. J. Biochem. 2001, 268, 1.11121095 10.1046/j.1432-1327.2001.01869.x · doi ↗ · pubmed ↗
- 4Y. Yin , E. Morgunova , A. Jolma , E. Kaasinen , B. Sahu , S. Khund‐Sayeed , P. K. Das , T. Kivioja , K. Dave , F. Zhong , K. R. Nitta , M. Taipale , A. Popov , P. A. Ginno , S. Domcke , J. Yan , D. Schübeler , C. Vinson , J. Taipale , Science 2017, 356, eaaj 2239.28473536 10.1126/science.aaj 2239 PMC 8009048 · doi ↗ · pubmed ↗
- 5S. Li , Y. Peng , A. R. Panchenko , Curr. Opin. Struct. Biol. 2022, 75, 102430.35914496 10.1016/j.sbi.2022.102430 · doi ↗ · pubmed ↗
- 6P. Koivunen , T. Laukka , Cell. Mol. Life Sci. 2018, 75, 1339.29184981 10.1007/s 00018-017-2721-8PMC 11105636 · doi ↗ · pubmed ↗
- 7a) F. Zhang , J. H. Pomerantz , G. Sen , A. T. Palermo , H. M. Blau , Proc. Natl. Acad. Sci. 2007, 104, 4395;17360535 10.1073/pnas.0700181104 PMC 1838613 · doi ↗ · pubmed ↗
- 8J. C. Price , E. W. Barr , L. M. Hoffart , C. Krebs , J. M. Bollinger , Biochemistry 2005, 44, 8138.15924433 10.1021/bi 050227 c · doi ↗ · pubmed ↗