Cys–Lys stapling for unprotected peptides via tunable linkers
Kaizhen Miao, Bei Fu, Leiyang Bai, Chengliang Li, Xuefeng Jiang

TL;DR
This paper introduces a new method for creating stable peptides by crosslinking cysteine and lysine using tunable linkers, improving their structure and function for drug development.
Contribution
A novel non-symmetric stapling method for Cys–Lys crosslinking in peptides using tunable linkers with high selectivity and self-assembly.
Findings
A library of 17 stapling reagents was developed with adjustable properties for Cys–Lys ligation.
The method enables macrocyclization of peptide loops into 25–40-membered rings under biocompatible conditions.
Stapled peptides showed improved helicity, stability, and anti-bladder cancer activity.
Abstract
Stapling has emerged as a transformative paradigm in peptide chemistry, enabling precise conformational control to endow peptides with augmented biophysical properties, including enhanced proteolytic stability and target-binding affinity. Although symmetric macrocyclization strategies have been investigated, non-symmetric stapling of native peptide scaffolds remains underexplored, owing to intricate synthetic challenges associated with achieving concurrent chemoselectivity and site selectivity. This limitation primarily stems from the requirement for orthogonal reactivity in modifying distinct proteinogenic residues while preserving native side-chain functionalities under biocompatible conditions. Herein, a non-symmetric stapling is disclosed for cysteine–lysine (Cys–Lys) crosslinking in unprotected peptides and proteins via unsymmetrically tunable linkers with high chemoselectivity and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
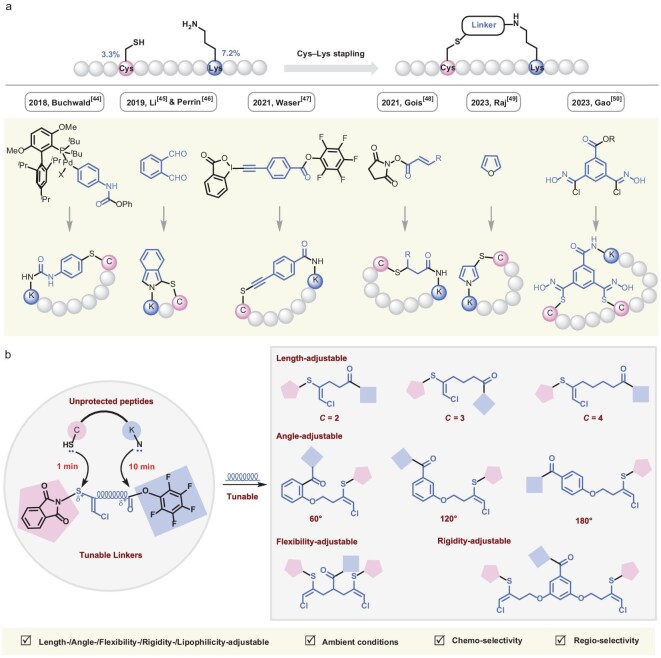 Figure 1
Figure 1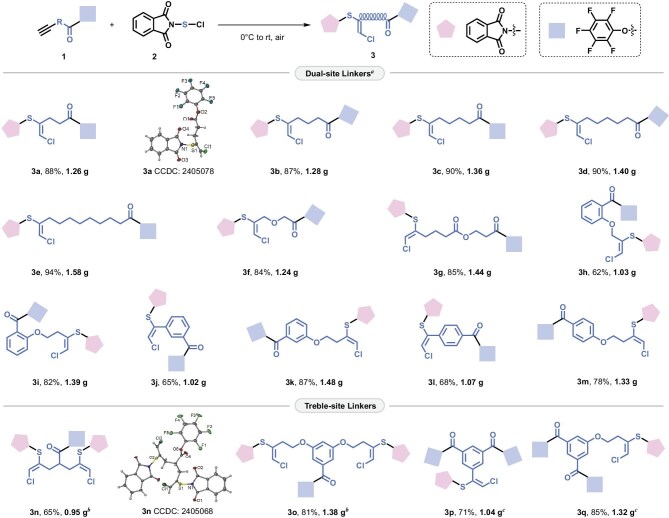 Figure 2
Figure 2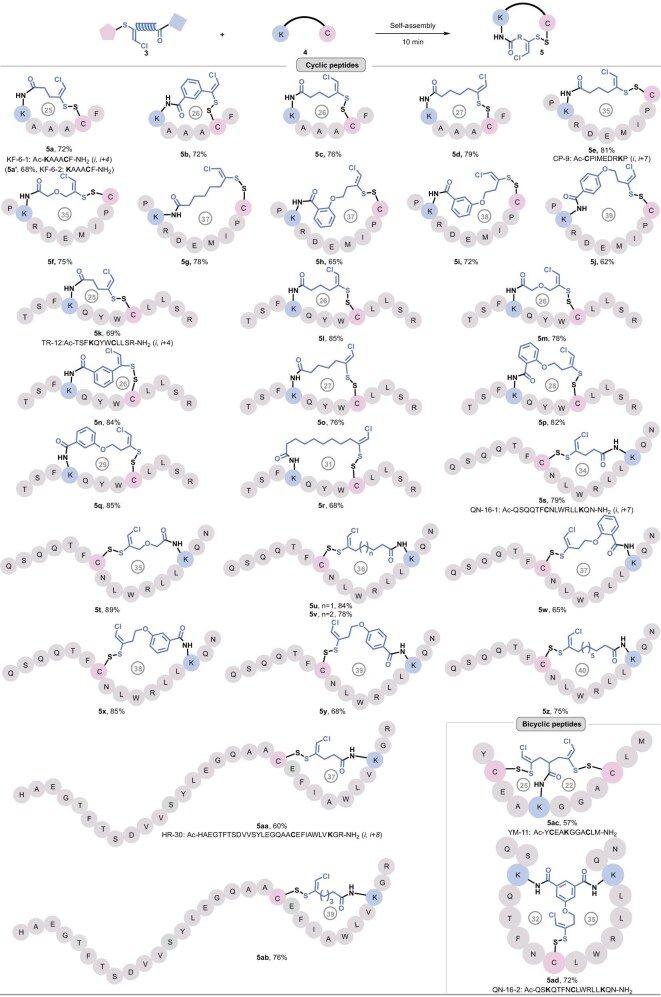 Figure 3
Figure 3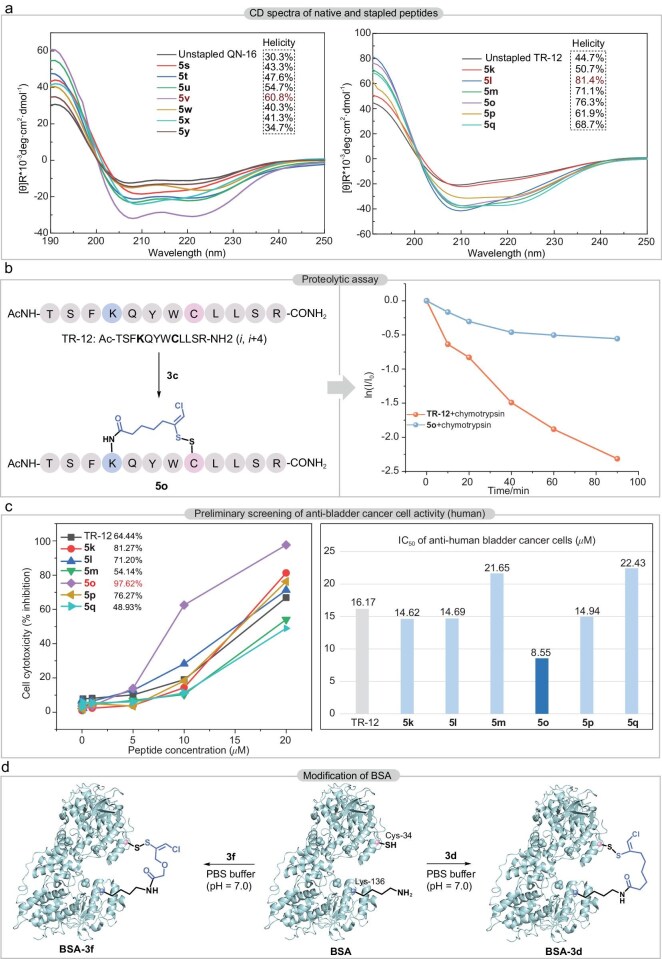 Figure 4
Figure 4- —National Natural Science Foundation of China10.13039/501100001809
- —Shanghai Rising-Star Program10.13039/501100013105
- —Science and Technology Commission of Shanghai Municipality10.13039/501100003399
- —China Postdoctoral Science Foundation10.13039/501100002858
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced biosensing and bioanalysis techniques · Chemical Synthesis and Analysis · Monoclonal and Polyclonal Antibodies Research
INTRODUCTION
Peptides have emerged as pharmacologically potent agents characterized by exquisite target specificity and enhanced safety profiles compared to conventional small-molecule therapeutics, driving their escalating clinical adoption across diverse therapeutic domains [1,2]. As of 2020, over 100 peptide-based compounds have received regulatory approval for therapeutic or diagnostic applications, underscoring their translational potential [3]. However, peptides are facing three major pharmacological challenges: (i) strong susceptibility to proteolytic degradation in vivo; (ii) poor membrane permeability; and (iii) low solubility in aqueous solutions, severely limiting their therapeutic efficacy [4,5]. Peptide stapling has proven ability for transforming linear peptides into conformationally constrained peptides, enhancing multiple properties of peptides (α-helix conformation, cell penetration, proteolytic stability, target selectivity, aqueous solubility, etc.) for drug development [6–8]. Building upon the seminal work of Blackwell and Grubbs [9], Verdine and co-workers [10,11] first introduced the term ‘stapled peptides,’ which has inspired multifaceted endeavors in peptide and protein stapling [12–15]. The stapling was typically mediated by symmetric linkers that crosslinked two identical native residues within the peptide sequence, thereby ensuring stereochemical uniformity and avoiding regioisomers. Representative examples have included the cysteine–cysteine (Cys–Cys) sulfhydryl stapling, [16–29] lysine–lysine (Lys–Lys) N-terminus stapling [23,30–33] and tyrosine–tyrosine (Tyr–Tyr) stapling [34,35] of native peptides. Unlike symmetric stapling, its non-symmetric counterpart relies on unsymmetric crosslinkers to conjugate two different native residues within a peptide sequence, carrying multidimensional topologies and functionalities that are in high demand in peptide drug discovery [36–38]. However, non-symmetric locking is confronted with formidable challenges: (i) two residue-specific modifications lead to much more complexity; (ii) peptides bearing multiple identical residues demand higher orthogonality; (iii) simultaneously modifying distinct proteinogenic residues requires higher chemoselectivity and site selectivity; (iv) designing unsymmetric linkers needs to balance the reactivities and compatibilities for two terminals; and (v) undesired side pathways prove competitive, chiefly among peptide oligomerization, degradation of the staple itself, and the generation of regioisomeric products. The strong nucleophilicity of their side chains (thiol in cysteine and amine in lysine), coupled with their distinct natural abundances (3.3% and 7.2%, respectively), makes cysteine (Cys, C) and lysine (Lys, K) predominant targets for chemospecific and site-specific protein modification [39–42]. The challenging non-symmetric Cys–Lys sulfhydryl/N-terminus stapling has been achieved via combining Cys arylation with Lys amidation by Kubota et al. [43], via o-phthalaldehyde-mediated intermolecular crosslinking of Cys and Lys by Zhang et al. [44] and Todorovic et al. [45], respectively, via hypervalent iodine reagents for Cys initiation and pentafluorophenyl (PFP) ester for Lys handles by Ceballos et al. [46], via N-hydroxysuccinimide-activated acrylic ester for Cys bioconjugation and Lys activation by Silva et al. [47], via a bis-aldehyde species generated in situ through furan oxidation by Wang et al. [48], via leveraging bis-chlorooxime–Cys bioconjugation and Lys amidation by Chen et al. [49] and in a stepwise manner [50,51] (Fig. 1a). In continuation of our effort on organosulfur chemistry [28,52–59], we designed bilateral unsymmetrical Cys–Lys stapling reagents for non-symmetric Cys–Lys crosslinking with unprotected peptides (Fig. 1b). Based on the hard/soft acids and bases principle [60], Cys preferentially crosslinked at the sulfur site leaving the phthalimide group in 1 min, whereas Lys favored amidation targeting at PFP ester in 10 min. The length, angle, flexibility, rigidity and lipophilicity of unsymmetrical linkers are readily adjustable for enhancing helical conformation, solubility, hydrolytic stability, membrane permeability, binding affinity and biological activity. The matching ligation speed of the Cys and Lys labeling process enables the orthogonal crosslinking under ambient conditions with high chemoselectivity and regioselectivity in a self-assembly manner [61,62], successfully constructing diverse monocyclic/bicyclic peptides (Fig. 1b).
Non-symmetric Cys–Lys stapling. (a) Previous strategies. (b) This work: Cys–Lys relay stapling in one step for unprotected peptides via tunable linkers.
RESULTS AND DISCUSSION
Library of unsymmetrical Cys–Lys stapling linkers
The alkyne-substituted PFP ester 1 (see Fig. 2) was initially assembled via a condensation reaction between commercially available alkynoic acids and perfluorophenol. The alkynyl ester 1 retained terminal alkynyl sites for the second addition with phthalimidosulfenyl chloride 2 under an ambient atmosphere [28], generating highly E-selective electrophilic sulfur functional group 3. The difunctional reactions established a series of bilaterally unsymmetrical reagents 3, in which readily available alkynoic acids enabled the adjustable length, angle, flexibility, rigidity and lipophilicity. As further represented in Fig. 2, linker 3a, featuring an ethyl linkage, was synthesized with a good yield and its E-configuration was unambiguously confirmed by X-ray crystallographic analysis. To incorporate adjustable length and flexibility, we designed extended linkers 3b–3e, which were synthesized with excellent yield on a gram scale. The O-embedded 3f and ester-embedded 3g further promoted the diversiform stapling with tunable lipophilicity. Aryl moieties were installed to adjust the rigidity and angle of linkers, in which o-, m- and p-substituted modes were well compatible (3h–3m). In addition to dual-site staples, we also developed trifunctional linkers (Cys–Cys–Lys, 3n–3o; Lys–Lys–Cys, 3p–3q), establishing a promising platform for constructing bicyclic peptides. X-ray study of 3n confirmed one PFP ester site and two electrophilic sulfur sites with E-configuration. Density functional theory (DFT) calculations gained further insight into the Z/E selectivity for stapling reagents (Fig. S1 in the Supplementary data). The nucleophilic addition from ethylethyne to phthalimidosulfenyl chloride generated a cyclopropenylthionium ion Int-6, spontaneously discharging a chloride ion. It triggered the ring opening via attack at the site with less steric hindrance (TS-7), delivering a thermodynamically stable E-configuration. All stapling reagents exhibited excellent solubility in acetonitrile and dimethylformamide (DMF) with concentrations ranging from 34 to 230 mM (Fig. S4). Although they demonstrated limited solubility in water, they were found to be soluble in the reaction system—a mixed solvent of acetonitrile and water (3 : 1, v : v)—ensuring the efficient proceeding of the stapling reaction.
Library of unsymmetrical Cys–Lys stapling linkers. aReaction conditions: 1 (3.0 mmol), phthalimidosulfenyl chloride 2 (3.3 mmol), CH2Cl2 (0.1 M), 0°C to rt, air, 2–12 h. All yields are for the isolated products. bReaction conditions: 1 (2.0 mmol), 2 (4.4 mmol). cReaction conditions: 1 (2.0 mmol), 2 (2.2 mmol).
Unsymmetrically stapled cyclic peptides
Subsequently, unsymmetrical peptide stapling was comprehensively explored (Fig. 3). Stapling reagent 3a crosslinked with unprotected peptide KF-6-1: Ac-KAAACF-CONH_2_ (i, i + 4) via Cys connection within 1 min under ambient conditions; subsequent Lys ligation was achieved with addition of N,N-diisopropylethylamine (DIPEA) for 10 min. The relay stapling cooperatively generated the cyclic peptide 5a with 72% yield. Notably, the native peptide KF-6-2: KAAACF-CONH_2_ without N-terminal acetylation protection was launched for the stapling process, generating 5a′ successfully, with 68% yield. It is worth noting that the fast disulfide click process was quantitative and specific, thereby enabling a one-step dual-stapling protocol within the same yield via adding 3a and DIPEA together to the peptide KF-6-1. Following the single-step method, 5b–5d were efficiently obtained in 72%–79% yields. The observed high chemoselectivity and regioselectivity were demonstrated via the control experiment of CP-9: AcHN-CPIMEDRKP-CONH_2_ (i, i + 7) and reagent 3b (Figs S5–S10). The mass spectrometry fragmentation analysis unambiguously evidenced that the specific crosslinking at the sulfur site was launched by Cys rather than by Lys (Figs S9 and S10). The reagent 3d with single Cys [(tert-butoxycarbonyl)-d-cysteine] (Figs S11–S15) and single Lys [tert-butyl (tert-butoxycarbonyl)-l-lysinate] (Figs S16–S19) further proved that Cys preferentially crosslinked with the sulfur site leaving the phthalimide group, whereas Lys favored amidation targeting at the PFP ester. Additionally, CP-9 was readily stapled by diversely functional linkers generating constrained peptides 5e–5j in elegant conversions, such as the o-substituted 5f (75%) and aryl-modified 5h–5j (62%–72%). The o-, m- and p-linked manner brought distinct rigidity and angle for binding the α-helix conformation (Fig. 3). The versatility of this approach was further demonstrated through the stapling of peptide TR-12 (Ac-TSFKQYWCLLSR-CONH_2_) at (i, i + 4) positions, successfully generating 25–31-membered macrocyclic peptides 5k–5r in yields ranging from 68% to 85%. Furthermore, more linkers with diverse length, angle, flexibility, rigidity and lipophilicity were assembled on peptide QN-16-1: Ac-QSQQTFCNLWRLLKQN-CONH_2_ (i, i + 7), delivering structurally locked peptides 5s–5z with 35- to 40-membered loops with satisfactory yields. Building on the success of dual-site crosslinking, we next applied the Cys–Cys–Lys reagent 3n and Lys–Lys–Cys reagent 3q to triple-site stapling, successfully generating bicyclic peptide architectures. To address the potential limitations of peptide length in this stapling methodology, a linear 30-mer peptide HR-30 was stapled with reagents 3a and 3c, successfully generating the target 37- and 39-membered macrocyclic peptides 5aa and 5ab with 60% and 76% isolated yields, respectively. Under fixed conditions with stapling reagent 3d, peptides KF-6-1, TR-12, QN-16-1 and HR-30 with varying lengths (6‒30-mer) delivered target stapled counterparts 5d, 5o, 5u and 5ab with good yields (76%‒84%), demonstrating that the stapling protocol exhibits broad applicability across diverse peptide sizes. Notably, the unprotected peptides YM-11 (Ac-YCEAKGGACLM-CONH_2_) and QN-16-2 (Ac-QSKQTFNCLWRLLKQN-CONH_2_) underwent smooth triple ligation. This transformation effectively locked the peptides into preorganized, rigid conformations, generating the respective bicyclic structures 5ac and 5ad (Fig. 3). Bicyclic peptides demonstrated superior properties over their monocyclic counterparts, including enhanced structural rigidity, improved target binding affinity and selectivity, greater metabolic stability and superior membrane permeability. [33,49,63,64]. The efficient relay conjugation highlights the orthogonal reactivity designed within the linkers: the phthalimide (PhthN^−^) group, with its superior leaving ability, provided a rapid and specific click site for highly nucleophilic Cys residues, whereas the PFP esters could only be activated by the higher nucleophilic Lys residues with the assistance of DIPEA, enabling the efficient construction of a library of multi-disulfide 25- to 40-membered macrocyclic peptides under biocompatible conditions.
Unsymmetrically stapled cyclic peptides prepared by preparative HPLC. Reaction conditions: 4 (3 or 5 µmol), 3 (1.5 equiv.), CH3CN:H2O = 3:1, rt, air, DIPEA (4.0 equiv.), 10 min.
Biological characterization
The circular dichroism (CD) spectrum [65] of 5y was measured in six different solvent systems (H_2_O, 5% trifluoroethanol (TFE)/H_2_O, 10% TFE/H_2_O, 20% TFE/H_2_O, 30% TFE/H_2_O and 40% TFE/H_2_O), in which the optimum α-helicity of 34.7% was observed in 40% TFE/H_2_O (Fig. S20). Two linear peptides TR-12 and QN-16 were contrasted with their stapled form under this condition (Fig. 4a). Compared to the 30.3% helicity of native peptide QN-16, the best helicity of its stapled peptide was 5v, reaching 60.8%. A similar reinforcement of the helicity was observed between TR-12 (44.7%) and its stapled peptide 5l (81.4%). Collectively, the data demonstrated that our stapling strategy reinforced the native α-helical fold, validating the rational design of the linkers. To evaluate the proteolytic stability of the stapled peptide [66], TR-12 and stapled peptide 5o were exposed to chymotrypsin (Fig. 4b). High-performance liquid chromatography (HPLC) analysis indicated that under chymotrypsin for 1.5 h, 90% of linear peptide degraded, in contrast with 43% of 5o. These results revealed an obviously enhanced proteolytic stability for stapled peptide compared to that of the unstapled precursor. To evaluate the bovine serum stability of stapled peptides, three representative linear peptide-derived binding peptides 5h, 5o and 5w were selected for comprehensive analysis under physiologically relevant conditions (37°C) (Figs S24–S26). CP-9-derived 5h demonstrated exceptional stability, requiring 14 h for complete degradation, while it took 8 and 6 h to fully decompose for TR-12-modified 5o and QN-16-engineered 5w, respectively. The differential stability patterns observed among the three architectures demonstrated structure-dependent resistance to enzymatic degradation in serum environments. Activity tests at a concentration of 20 μM showed that peptide TR-12 had an inhibitory effect on human bladder cancer cells (64.44%) (Fig. 4c). We conducted anti-bladder cancer activity screening, in which stapled peptides 5k (81.27%), 5l (71.20%), 5o (97.62%) and 5p (76.27%) showed superior inhibitory activity. Additionally, the activity of the stapled peptides 5m (54.14%) and 5q (48.93%) were not compromised either. Furthermore, half-maximal inhibitory concentration (IC_50_) assays indicated that the constrained peptides exhibited increased IC_50_ values: TR-12 (16.17 μM), 5k (14.62 μM), 5l (14.69 μM), 5m (21.65 μM), 5o (8.55 μM), 5p (14.94 μM) and 5q (22.43 μM), which was consistent with the inhibitory activity (Fig. 4c). The observed disparity between the native and unstapled peptides served as direct validation of the strategy’s superiority. Furthermore, the amino-sulfhydryl stapling of bovine serum albumin (BSA) was performed, a protein with a molecular weight of 66 kDa containing 59 lysine residues and a single free cysteine residue (Cys-34, Fig. 4d). The thiol group of cysteine (Cys-34) and amine group of lysine residues in BSA were stapled through reagents 3d and 3f under the conditions of PBS buffer (pH 7.0) and the presence of DIPEA with a protein concentration of 50 μM within 10 min. MALDI-TOF MS analysis revealed characteristic molecular weights of the modified protein (67274.58 Da for BSA-3d, Fig. S29; and m/z = 67248.66 Da for BSA-3f, Fig. S30), demonstrating the biocompatibility of this stapling methodology in biological environments. Based on the amino acid sequence of BSA, we predicted its structure using AlphaFold2 and visualized the distribution of lysine residues proximal to Cys residues in VMD software. Given that the chain length of the stapling reagent used here is similar to that employed by the Gois group in their BSA stapling [67], we hypothesized that BSA underwent stapling with the reagent at Cys34 and Lys136 (Fig. S31).
(a) CD spectra of native and stapled peptides in 40% TFE/water. (b) Proteolytic assay of stapled peptide 5o and its native precursor TR-12. (c) Biological characterization of anti-human-bladder cancer cells. Three independent experiments were conducted, and the results were normalized. (d) Modification of BSA with 3d and 3f in PBS buffer (50 mM, pH = 7.0), rt, air, DIPEA (10.0 equiv.), 10 min.
CONCLUSION
We have demonstrated Cys–Lys relay non-symmetric stapling with unprotected peptides/protein via a 17-membered library of unsymmetrical linkers with high chemoselectivity and regioselectivity in a self-assembly manner, stitching 6–30 amino acid residues to generate 25- to 40-membered conformationally restricted peptides in biocompatible media. The tunable geometry, including length, angle, flexibility and rigidity of stapling reagents, manipulated the crosslinking efficiency, cooperatively enhancing the pharmacological properties, such as α-helicity, proteolytic stability, serum stability and anti-bladder cancer activity. This protocol can be further applied for the establishment of various functional cyclic/bicyclic peptide libraries in both chemical biology study and peptide-based drug discovery.
Supplementary Material
nwaf406_Supplemental_Files
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Muttenthaler M, King GF, Adams DJ et al. Trends in peptide drug discovery. Nat Rev Drug Discov 2021; 20: 309–25.10.1038/s 41573-020-00135-833536635 · doi ↗ · pubmed ↗
- 2Liao Y, Jiang X. Chemo-selective modification of cysteine residue: synthesis and application in the discovery of potential drug candidates. Explor Drug Sci 2024; 540–54.10.37349/eds.2024.00060 · doi ↗
- 3D’Aloisio V, Dognini P, Hutcheon GA et al. Pep Ther Dia: database and structural composition analysis of approved peptide therapeutics and diagnostics. Drug Discov Today 2021; 26: 1409–19.10.1016/j.drudis.2021.02.01933647438 · doi ↗ · pubmed ↗
- 4Tyndall DA, Nall T, Fairlie D. Proteases universally recognize beta strands in their active sites. Chem Rev 2005; 105: 973–1000.10.1021/cr 040669 e 15755082 · doi ↗ · pubmed ↗
- 5Jenssen H, Aspmo S. Serum stability of peptides. Methods Mol Biol 2008; 494: 177–86.18726574 10.1007/978-1-59745-419-3_10 · doi ↗ · pubmed ↗
- 6Walensky LD, Bird GH. Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem 2014; 57: 6275–88.10.1021/jm 4011675 PMC 413668424601557 · doi ↗ · pubmed ↗
- 7Vinogradov AA, Yin Y, Suga H. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J Am Chem Soc 2019; 141: 4167–81.10.1021/jacs.8b 1317830768253 · doi ↗ · pubmed ↗
- 8Colas K, Bindl D, Suga H. Selection of nucleotide-encoded mass libraries of macrocyclic peptides for inaccessible drug targets. Chem Rev 2024; 124: 12213–41.10.1021/acs.chemrev.4c 00422 PMC 1156557939451037 · doi ↗ · pubmed ↗