Effects of avian Plasmodium exposure on the microbiota of Culex pipiens
Marta Garrigós, Olaya García-Ruiz, Charlotte R. Enkvist, María José García-López, Isabel Moreno-Indias, María José Ruiz-López, Jesús Veiga, Jordi Figuerola, Elin Videvall, Josué Martínez-de la Puente

TL;DR
This study explores how exposure to avian malaria parasites affects the gut bacteria of a mosquito species, finding subtle changes in bacterial composition.
Contribution
The study investigates the impact of avian Plasmodium on Culex pipiens microbiota using a 16S rRNA metabarcoding approach.
Findings
Exposure to avian Plasmodium increased relative abundance of Bacteroidaceae and Bacteroides in mosquito microbiota.
Excluded Wolbachia, exposed mosquitoes showed higher Rikenellaceae abundance.
Bacterial communities in exposed mosquitoes were enriched in biosynthesis and metabolism pathways.
Abstract
Malaria parasites (Plasmodium spp.) are mosquito-borne parasites that infect humans and wildlife. Several studies support the role of mosquito microbiota as a major driver of Plasmodium transmission, although studies on wildlife malaria are typically neglected. Here, we used a 16S rRNA metabarcoding approach to assess whether the exposure to avian Plasmodium parasites affects the microbiota of their natural vector, Culex pipiens. Mosquitoes, captured in the field as larvae and grown in the laboratory, were allowed to feed on house sparrows (Passer domesticus) naturally infected with Plasmodium relictum (lineage SGS1) and uninfected birds. We analyzed the microbiota composition of the abdomens of individual mosquitoes and found 2,006 Amplicon Sequence Variants (ASVs). Culex pipiens’ microbiota was dominated by bacteria of the genus Wolbachia, followed by the genera Stenotrophomonas and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
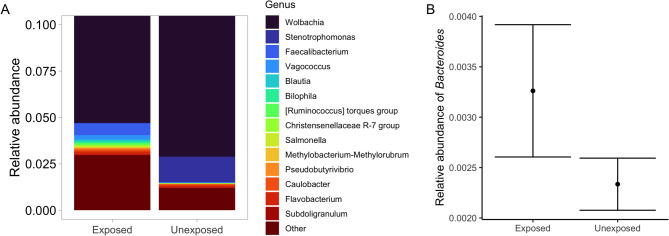 Figure 1
Figure 1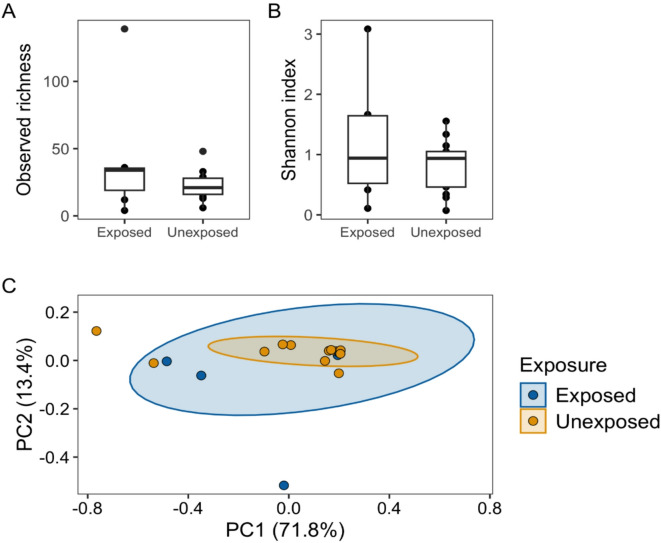 Figure 2
Figure 2- —https://doi.org/10.13039/100014440Ministerio de Ciencia, Innovación y Universidades
- —https://doi.org/10.13039/501100011033Agencia Estatal de Investigación
- —https://doi.org/10.13039/501100004587Instituto de Salud Carlos III
- —Swedish Research Council
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect symbiosis and bacterial influences · Bird parasitology and diseases · Invertebrate Immune Response Mechanisms
Introduction
Malaria, caused by parasites of the genus Plasmodium, produces around half a million human fatalities annually^1^. In addition to humans, other non-human vertebrates are infected by Plasmodium parasites, causing diseases with ecological and evolutionary consequences^2^. Most Plasmodium parasites require mosquitoes to complete their life cycle and be transmitted from an infected individual to a new host. The development of Plasmodium in mosquitoes is modulated by abiotic and biotic factors, including intrinsic factors such as the composition of the digestive tract bacterial microbiota of mosquitoes (hereafter gut microbiota)^3,4^. Mosquitoes acquire most of their gut microbiota during the aquatic larval stage from their breeding sites^4^, while a small proportion of this microbiota is transferred to the adult stage through transstadial transmission^5^. The gut microbiota of adults is modified by their feeding sources, including plant sugars, water and, in the case of females, the blood meals from vertebrate hosts^6,7^.
The mosquito microbiota interacts with Plasmodium through direct mechanisms, producing metabolites with antiparasitic properties and reactive oxygen species^8^, but also through indirect mechanisms. The microbiota not only enhances the activation of mosquito immune responses, it also favors the formation of physical barriers against the parasite (e.g., the gut peritrophic matrix)^9^. Disruptions of the microbiota affect the susceptibility of mosquitoes to Plasmodium, as supported by studies with different malaria models^10,11^. However, contradictory results have been found depending on the bacteria-parasite-mosquito system studied. For example, bacteria of the genera Comamonas, Acinetobacter, Pseudomonas, Pantoea, Serratia, Elizabethkingia,* Enterobacter* and Chromobacterium totally or partially blocked the development of Plasmodium falciparum in Anopheles gambiae^8,12,13^. Wolbachia increases the susceptibility of Culex pipiens mosquitoes to Plasmodium relictum, by increasing the prevalence of the parasite’s infective stage^14^. On the other hand, Plasmodium infection can also affect the microbiota composition of mosquitoes. Aželytė et al.^15^ found significant differences in the relative abundance of 20 bacterial genera between P. relictum-infected and uninfected Culex quinquefasciatus mosquitoes, using mosquito colonies and experimentally infected canaries as parasite donors. However, further studies are necessary to reveal the interactions between mosquito microbiota and avian Plasmodium, especially using natural parasite-mosquito assemblages, given the strong impact that the environment may have on the microbiota composition of mosquitoes^16,17^.
Plasmodium relictum is a generalist malarial parasite that infects bird species worldwide and is considered one of the most dangerous invasive species due to its negative effects on bird populations in invaded areas^2,18^. Culex pipiens is a well-known vector of avian Plasmodium, including P. relictum^19^. Here, we performed a proof-of-concept study to identify the potential associations between mosquito microbiota and exposure to avian Plasmodium using a natural host-parasite-vector assemblage. Using a 16S rRNA metabarcoding approach, we compared the abdominal microbiota (hereafter referred to as microbiota in the context of this study) of Cx. pipiens females 21 days after feeding on *P. relictum-*infected house sparrows with that of females fed on uninfected sparrows.
Results
We initially selected 64 mosquitoes to be analyzed for microbiota composition. There was no amplification of the 16S rRNA gene for five samples and fewer than 1,000 reads were obtained for 39 samples. Therefore, after the data filtering steps, 20 samples were included in the final analysis, corresponding to 13 unexposed mosquitoes (fed on uninfected birds) and seven exposed mosquitoes (fed on birds infected by P. relictum - lineage SGS1). Two of the seven exposed mosquitoes tested positive for parasite infection in the head and thorax, and five were negative.
The analyses of the microbiota of these 20 Cx. pipiens resulted in a total of 6,911,775 reads, ranging from 1,134 to 1,816,458 (average = 345,588.75). Overall, 2,006 Amplicon Sequence Variants (ASVs) were identified, classified to 23 different phyla and, at least, 46 classes, 115 orders, 199 families, and 405 genera (Fig S1-S5). In terms of relative abundance, the microbiota of Cx. pipiens was dominated by the phylum Proteobacteria (average: 94.86%, SD = 0.124), followed by Bacillota (formerly Firmicutes) (average: 3.51%, SD = 0.110), Actinobacteriota (average: 0.79%, SD = 0.010), Cyanobacteria (average: 0.32%, SD = 0.006), and Bacteroidota (average: 0.31%, SD = 0.004) (Fig S1). Most reads (96.56%) corresponded to the genus Wolbachia (family Anaplasmataceae), which was the most abundant taxon in all samples (average relative abundance = 91.30%, SD = 0.141), followed by Stenotrophomonas (family Xanthomonadaceae), Faecalibacterium (family Ruminococcaceae), Vagococcus (family Enterococcaceae), Blautia (family Lachnospiraceae), Bilophila (family Desulfovibrionaceae), and Ruminococcus (family Ruminococcaceae), all of them with an average relative abundance below 2% (Fig. 1A) (see Supplementary Table S1 for the relative abundance of all genera).
Fig. 1. Abdominal microbiota of mosquitoes fed on *Plasmodium relictum-*infected (exposed) and uninfected birds (unexposed). (A) Relative abundance of the 15 most abundant genera of bacteria in exposed and unexposed mosquitoes. The y-axis shows a zoomed-in version of the relative abundance of bacterial genera from 0 to 0.1. From 0.1 to 1, the relative abundance corresponds solely to the Wolbachia genus (not pictured). (B) Relative abundance of the genus Bacteroides in the abdominal microbiota of exposed and unexposed mosquitoes. Bars correspond to standard errors.
Fig. 2. Diversity of the abdominal microbiota of mosquitoes fed on *Plasmodium relictum-*infected (exposed) and uninfected birds (unexposed). (A) Distribution of observed richness in the microbiota of exposed and unexposed mosquitoes. The horizontal lines represent the median observed richness of each group. The boxes delimit the upper (75%) and lower (25%) quartiles, and the vertical lines go from the lower and upper quartiles to the minimum or maximum values. (B) Distribution of the Shannon index estimate in the microbiota of exposed and unexposed mosquitoes. (C) Principal Co-ordinates Analysis (PCoA) for Bray-Curtis distance matrix (relative abundance) in the microbiota of exposed and unexposed mosquitoes. PC1 and PC2 axes refer to the percentage of variation explained by each of the two selected main coordinate axes.
We did not find significant differences in the alpha diversity of the microbiota between exposed and unexposed mosquitoes, including richness (Kruskal–Wallis (K–W): χ² = 1.146, Degrees of freedom (Df) = 1, P = 0.284) and Shannon index (K–W: χ² = 0.566, Df = 1; P = 0.451) (Fig. 2A and B). Similarly, we did not find significant differences in the beta diversity of the microbiota of exposed and unexposed mosquitoes (PERMANOVA: F = 0.381, Df = 1, P = 0.77) (Fig. 2C). The microbiota of exposed and unexposed mosquitoes shared a total of 126 families (63.32%) and 204 (50.37%) genera, with the remaining bacteria being exclusively found in one of the two experimental groups (Fig S6; Supplementary Table S2). Moreover, a differential abundance analysis showed that the microbiota of exposed mosquitoes had a higher relative abundance of the family Bacteroidaceae (ancombc2: Log Fold Change (LFC) = 3.14; P = 0.014) and the genus Bacteroides (ancombc2: LFC = 3.14; P = 0.024) than that of unexposed mosquitoes (Fig. 1B).
We repeated the analyses after excluding ASVs belonging to the genus Wolbachia. After filtering for number of reads, this new dataset included 18 mosquitoes (13 unexposed and five exposed). The results were similar to those obtained when Wolbachia was included in the analyses. Again, no differences were found in the alpha diversity, including richness (K–W: χ² = 2.1885, Df = 1, P = 0.139) and Shannon index (K–W: χ² = 1.7709, Df = 1, P = 0.183) (Fig S7A and B), and beta diversity (PERMANOVA: F = 0.658, Df = 1, P = 0.87), between the microbiota of exposed and unexposed mosquitoes (Fig S7C). However, the microbiota of exposed mosquitoes had a higher relative abundance of the bacterial family Bacteroidaceae (ancombc2: LFC = 2.82; P = 0.019) and the genus Bacteroides (ancombc2: LFC = 2.82; P = 0.034) than that of unexposed mosquitoes. In addition, the microbiota of exposed mosquitoes also had a higher relative abundance of the family Rikenellaceae (ancombc2: LFC = 1.93; P = 0.048).
A total of 7,178 Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs were identified from the bacterial community of Cx. pipiens. The KEGG pathway analysis revealed that the microbiota of exposed mosquitoes differed in 70 pathways (|LFC| > 2, 0.98%) compared with the microbiota of unexposed mosquitoes. These pathways were related to ribosome, two-component system, ABC transporters, calcium signaling pathway, focal adhesion, oxidative phosphorylation, peptidoglycan biosynthesis, and tryptophan metabolism, among others (Supplementary Table S3). After excluding Wolbachia, the microbiota of exposed mosquitoes differed in 122 pathways (1.70%). These 122 differentially abundant pathways included all pathways previously found when Wolbachia was considered in the analysis, together with pathways related to metabolism of different biomolecules like galactose, starch and sucrose, D-amino acid, lysine, cysteine and methionine, and pyruvate. Additional differentially abundant pathways included the phosphotransferase system (PTS) and DNA replication, among others (Supplementary Table S3).
Discussion
The characterization of the abdominal microbiota of adult Cx. pipiens mosquito females emerged in the laboratory from field-collected larvae showed the dominance of the phylum Proteobacteria. Other bacteria of the phyla Bacillota, Bacteroidetes, Actinobacteria, and Cyanobacteria were also commonly found, as previously reported in studies using wild mosquito populations^6,20^. Wolbachia was the most abundant genus in the samples representing, on average, more than 90% of the mosquito abdominal microbiota. This bacterium typically dominates the general microbial community of many wild mosquitoes, including Cx. pipiens^21–23^. Wolbachia spp. are endosymbiotic bacteria present in different mosquito tissues, with particularly high abundance in reproductive organs^24^, but also found in other tissues like the digestive tract^25^. The next most abundant genera in our samples were Stenotrophomonas, Faecalibacterium, Vagococcus, Blautia, Bilophila, and Ruminococcus. A similar bacterial community was previously found in whole-body Aedes albopictus samples from Madagascar, which was dominated by Wolbachia, but also presented bacteria of the genera Blautia, Faecalibacterium, and Ruminococcus^21^. These bacteria, together with Stenotrophomonas, are part of the microbiota of different mosquito species in other geographical areas^26–28^. The microbiota found in the related species Cx. perexiguus from southern Spain is, however, very different, because Wolbachia is virtually absent^29^. The differences in the microbiota composition between species from the same region support the relevance of mosquito species identity as a major driver of the composition of mosquito microbiota in the wild^22^. Because the microbiota of mosquitoes may vary between body parts, future studies focusing on the characterization of the gut microbiota may dissect the posterior midgut of mosquitoes, and extract DNA from this tissue alone, instead of using the whole abdomen, as we did.
We found a higher relative abundance of bacteria of the family Bacteroidaceae and the genus Bacteroides in the abdominal microbiota of exposed mosquitoes compared to unexposed ones. The genus Bacteroides has been associated with a higher susceptibility to Plasmodium in humans and rodents^30^. In mosquitoes, the bacterium Bacteroides vulgatus was enriched in wild Aedes mosquitoes infected by Zika virus^28^. Our results were highly consistent when both including and excluding Wolbachia from our dataset. In addition, we identified a higher relative abundance of the Rikenellaceae bacteria in exposed mosquitoes, when Wolbachia was excluded from the analyses. As a member of insect gut microbiota, the Rikenellaceae family plays a role in vitamin synthesis^31^, and is highly abundant in mosquitoes from, for instance, Brazil, especially during the larval stage^32^. However, to our knowledge, it has not been previously associated with Plasmodium parasites in mosquitoes. In addition to differences in mosquito microbiota, we also identified differentially abundant KEGG orthologs (KOs) between the microbiota of exposed and unexposed mosquitoes, obtaining similar results with and without Wolbachia. The microbiota of exposed mosquitoes showed an enrichment in pathways related to metabolism. Furthermore, despite minor whole-community differences in the microbiota of mosquitoes according to the experimental procedure, the microbiota of exposed mosquitoes was generally enriched with the two-component system, a signal transduction pathway in bacteria regulating responses to environmental stimuli^33^. These metabolic associations should be further explored by assessing the correlation between the mosquito microbiota and transcriptomic responses.
Due to the cross-sectional experimental approach, we were not able to determine if differences in mosquito microbiota were caused by or a consequence of avian Plasmodium exposure. However, the fact that mosquitoes exposed to infected and uninfected birds had the same origin, potentially harboring a similar microbiota, suggests that differences could potentially be caused by parasite exposure. Culex pipiens can develop infective parasite stages after feeding on birds with low Plasmodium parasitemia levels (diagnosed negative according to blood smear observation)^34^, although some exposed mosquitoes may remain uninfected^35^. Indeed, we only detected parasite DNA in two of the seven exposed mosquitoes. In addition, the pattern found here could partly reflect differences in the characteristics of avian blood meals from infected and uninfected birds. Red cell density, metabolites, and immune factors in the blood are likely to differ between infected and uninfected birds^36,37^, which can affect mosquito physiology^38^, potentially leading to shifts in the composition and function of the microbiota. These changes could affect mosquito resistance to pathogens, lifespan and fecundity^10^. Another source of variation in the mosquito microbiota could potentially be due to differences in the bacterial communities of the skin and feathers of birds used as donors, which might be ingested by mosquitoes during blood-feeding.
Finally, in spite of using organisms from the field including wild house sparrows naturally infected by the local avian Plasmodium strain and field-collected vectors as larvae, our results could be affected by the conditions used during the experiments. These include, for example, the exposure to bacteria found in the laboratory during mosquito development or the food sources used for their maintenance. This experimental setup is unlikely to exactly reflect the conditions found in the wild. In addition, including mosquitoes that received different concentrations of sugar in their diet could affect the results, although this variable was confirmed to not be significantly correlated with bacterial alpha and beta diversity in this study (Supplementary Material 1) and was corrected for in the differential abundance models.
Conclusion
Our results support a minor but significant difference in the Cx. pipiens mosquito abdominal microbiota after exposure to avian *Plasmodium-*infected birds, which could be detected in the metabolic pathways of bacteria. In order to confirm causality of this relationship, experimental manipulation of the mosquito microbiota, with a special focus on the family Bacteroidaceae, prior to the exposure to parasite infections, will be necessary.
Materials and methods
Experimental setup
Wild juvenile house sparrows (Passer domesticus) (n = 20) were captured with mist nets in August 31st and September 9th 2021, in Trigueros (Huelva province, Spain; 37°19’33.7"N, 6°47’45.6"W). Each house sparrow was ringed, weighed and the wing length measured before being brought into captivity to the animal facilities of the Doñana Biological Station. All procedures were conducted in accordance with relevant guidelines and regulations, including compliance with national legal requirements for animal experimentation (Article 34 RD 53/2013). Following the conclusion of the experiment, the birds were released at the original capture site. The protocol was approved by the Animal Experimentation Committee (ref. 12/07/219/129). Reporting of methods and results adheres to the ARRIVE guidelines. A blood sample was collected from the jugular vein of each bird using a sterile syringe to molecularly identify infection status and the parasite lineage identity. A drop of blood was used to make a blood smear. Genomic DNA was extracted from blood samples using Maxwell^®^ 16 LEV Blood DNA Kit (Promega, Madison, WI, USA)^39^ and the Plasmodium infection status and parasite identity were assessed following the protocol by Hellgren et al.^40^. This method has a sensitivity of, at least, one parasite per 100,000 host blood cells. The amplified products of the positive samples were sequenced on both strands using Capillary Electrophoresis Sequencing by Macrogen (Madrid, Spain). The sequences were analyzed using Geneious v. 2020.0.3^41^ and assigned to lineages using the blast tool in MalAvi^42^. The blood smears were used to visually check the infection status and to quantify the levels of parasitemia in each bird using a light microscope ZEISS Axioscope 5. We observed no parasite cells in 10,000 erythrocytes in uninfected birds and one mature parasite cell per 10,000 erythrocytes in infected birds. After molecular and microscopic analysis, six birds were chosen for further procedures: three birds infected by P. relictum lineage SGS1 and three uninfected birds. These birds were verified to be uninfected by other blood parasites of the related genera Haemoproteus and Leucocytozoon. At the end of the experiments, birds were blood sampled again to test the presence of infection and quantify the parasitemia levels. PCR tests confirmed that the three uninfected birds remained negative for avian malaria, and the three infected birds continued to test positive for Plasmodium. However, blood smears from the infected birds no longer presented parasites, suggesting that the intensity of parasitemia might have decreased to fewer than one parasite cell per 10,000 erythrocytes.
Mosquito larvae were collected in September and October of 2021 from a single pond in La Rinconada (Seville, province Spain; 37º 29′ 16.8′′ N, 5º 58′′ 44.4′′ W). Once in the laboratory, larvae were placed in plastic trays with dechlorinated water and fed ad libitum with Hobby-Mikrozell 20 ml/22 g (Dohse Aquaristik, Gelsdorf, Germany) and Hobby-Liquizell 50 ml (Dohse Aquaristik). Larvae and adult mosquitoes were maintained under controlled conditions (26 °C ± 1, 55–60% relative humidity (RH) and 12:12 light: dark photoperiod cycle). Recently emerged adult mosquitoes had access to 0.5 w/v% (0.5%) sucrose solution for a maximum of 24 h to increase their chances of survival^43^. Adult Cx. pipiens females were morphologically identified under a stereomicroscope following Schaffner et al.^44^. The females were then sorted into groups of equal numbers and placed into separate insect cages (BugDorm-43030 F, 32.5 × 32.5 × 32.5 cm) with three different sucrose concentrations: ad libitum access to 0.5%, 2% and 8% in the context of a related study (unpublished data). Cages were randomly placed at different positions in the climatic chambers to control for possible spatial bias. For each diet-group, seven-day-old female mosquitoes were allowed to feed overnight on either a *P. relictum-*infected bird or an uninfected bird. Each bird was placed in a cage containing approximately 100 mosquitoes. A minimum of eight days passed from bird capture (when parasite detection was recorded in peripheral blood samples) to mosquito exposure. Fully blood-engorged females were placed in separate cages according to the diet they received before and after the blood meal (0.5%, 2% or 8%) and the infection status of the bird they fed on (positive or negative). We used six different birds in this experiment, three *P. relictum-*infected and three uninfected birds. Each mosquito-diet group was exposed to a different pair of birds, consisting of one infected and one uninfected bird. We performed three replicates of this experiment; in each replicate, we rotated the bird pairs among the diet groups, so that all birds were exposed to mosquitoes from all sucrose diets. The mosquitoes included in the final analysis fed on five different house sparrows, including three Plasmodium-infected and two uninfected birds (Table 1). Mosquitoes were kept in the same climatic chamber for 21 days after feeding to allow parasite development until the sporozoite stage^45^. Mosquitoes were then frozen at -80 °C until molecular analyses.
Table 1. Total number of samples (N) included in the final analysis (after filtering by number of reads), according to replicate, bird ID, bird infection status, mosquito infection status and diet. N ReplicateBird IDBird infection statusMosquito infection statusDiet(% sugar)3A178Negative–0.53A182Negative–22C178Negative–84E178Negative–21A165PositiveNegative21C164PositiveNegative81E187PositiveNegative0.51E187PositivePositive0.52E164PositiveNegative21E164PositivePositive2*Groups for which one sample was removed in the analysis excluding Wolbachia.
Molecular and bioinformatic analysis
Mosquitoes were sterilized individually by submerging them three times in 70% ethanol and once in sterilized water. Similar approaches have been previously used^21^, although this method may not fully eliminate external DNA^46^. Subsequently, mosquitoes were dissected to separate the head and thorax from the abdomen using two sterilized pipette tips in a sterilized petri dish. Genomic DNA from the head and thorax of mosquitoes was extracted using the Maxwell^®^ 16 LEV Blood DNA Kit (Promega, Madison, WI). We analyzed the Plasmodium infection status of mosquitoes following Hellgren et al.^40^.
The abdomens of 64 mosquitoes were used for individual microbiota characterization. The DNA was extracted using the DNeasy Blood & Tissue Kit ^®^ (QIAGEN, Hilden, Germany). The DNA from each mosquito was then used to build microbiota sequence libraries using the Ion 16S Metagenomics kit (Thermofisher, Waltham, Massachusetts, USA), which consists of primer pools that amplify multiple variable regions (V2, 3, 4, 6–7, 8 and 9) of the 16S ribosomal RNA gene. After generating amplicons, the Ion PlusTM Fragment Library Kit (Thermofisher, Waltham, Massachusetts, USA) was used to ligate individual barcoded adapters and synthesize separate libraries for each sample. Barcoded libraries from all the samples were pooled and templated on the automated Ion Chef system (Thermofisher, Waltham, Massachusetts, USA), followed by a 400 bp sequencing on the Ion S5 (Thermofisher, Waltham, Massachusetts, USA). The quality of the reads was assessed using FastQC (ver. 0.12.1)^47^ and MultiQC (ver. 1.17)^48^. The following analyses were performed in R (ver. 4.4.1)^49^ using Bioconductor packages. The dada2 package (ver. 1.32.0)^50^ was used to filter and trim the raw reads, and to construct ASVs using the parameters for Ion Torrent data recommended by the developer (https://benjjneb.github.io/dada2/tutorial.html). The dada2 package was also used to assign each ASV to a taxonomic group based on the SILVA database (ver. 138.1)^51^. Reads were filtered, removing singletons, reads that lacked taxonomic assignment below phylum level, and non-bacterial, chloroplast, and mitochondrial ASVs using the phyloseq package (ver. 1.48.0)^52^. Samples with fewer than 1,000 reads were discarded. In addition, since bacteria of the genus Wolbachia are endosymbionts distributed across various mosquito tissues^24^, we repeated the analysis using the dataset excluding ASVs corresponding to this genus. In this additional analysis, we discarded samples with fewer than 500 reads, resulting in a final dataset of 18 individuals.
Statistical analysis
We first performed an exploratory analysis of the microbiota composition according to the infection status of the mosquitoes (Supplementary Material 1; Figs S8A, S8B and C). Then, we conducted the statistical analyses by treating mosquitoes as two experimental groups: mosquitoes fed on P. relictum-infected birds (exposed mosquitoes) and mosquitoes fed on uninfected birds (unexposed mosquitoes). Comparisons of the bacterial alpha and beta diversity between groups were conducted in R using packages phyloseq, vegan (ver 2.6-8)^53^, and microbiome (ver. 1.26.0)^54^. Bacterial alpha diversity (within-sample diversity) of ASVs was estimated using observed richness and the Shannon index on samples rarefied to the minimum number of reads present in the samples. The Shannon index accounts for both ASV richness and evenness within a sample, increasing when the number of taxa is higher and more evenly distributed^55^. The Shapiro test^56^ was used to confirm the lack of normality of both observed richness and Shannon index. Kruskal-Wallis tests^57^ were used to statistically test differences in both observed richness and Shannon index by experimental groups (exposed and unexposed mosquitoes). Beta diversity (between-sample diversity) was analysed by a Principal Coordinate Analysis (PCoA) of Bray-Curtis dissimilarity matrix on the ASVs’ relative abundances. PERmutational Multivariate ANalysis Of VAriance (PERMANOVA)^58^ was used to test for differences between the centroids of the experimental groups using adonis2 in vegan. Statistical significance of F-values obtained from the PERMANOVA was determined through 999 permutations. The same approaches were used to corroborate the lack of association between bacterial alpha and beta diversity and the variables diet and replicate (Supplementary Material 1). Differences in the relative abundance of different taxa (i.e., microbiota composition) according to experimental groups were assessed at the family and genus levels using the Analysis of Compositions of Microbiomes with Bias Correction 2 (ANCOM-BC2) methodology from the ancombc package (ver. 2.6.0)^59^. Briefly, this model consists of a differential abundance analysis of microbiome data through a log-linear regression model while accounting for sample-specific and taxon-specific biases and correcting false positive errors by multi-group comparisons and repeated measurements^59^. Taxa with a prevalence below 10% were filtered out. Holm’s method was used to adjust the p-values for multiple testing. Because mosquitoes in this study fed on different sugar concentrations (0.5%, 2%, and 8%) and originated from three independent replicates, we adjusted the ANCOM-BC2 model for these potential confounders by including them as fixed effects alongside the main variable of interest, parasite exposure. Plots were made using the ggplot2 package (ver. 3.5.1)^60^.
Additionally, for both datasets with and without Wolbachia, we used PICRUSt2 (ver. 2.5.2) to infer the functional profiles of the microbial communities in each sample based on KEGG annotations^61^, obtaining predicted abundances for each KO identifier. In R, we filtered out KOs with a total predicted abundance below 10 across all samples, and compared the predicted abundances between exposed and unexposed mosquitoes using the DESeq2 package (ver. 1.44.0)^62^. According to the ancombc2 analysis, we adjusted the DESeq2 model for diet and replicate by including them as fixed effects. KOs with a p-value < 0.05 and a |LFC| > 2 were considered differentially abundant between groups. Finally, we retrieved the metabolic pathways associated with each significant KO using the KEGGREST package (ver. 1.44.1)^63^.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1.
Supplementary Material 2.
Supplementary Material 3.
Supplementary Material 4.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1WHO. World malaria report 2023: Tracking progress and gaps in the global response to malaria. In World Health Organization. (2023). https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2023.
- 2Muturi, E. J., Dunlap, C., Ramirez, J. L., Rooney, A. P. & Kim, C. H. Host blood-meal source has a strong impact on gut microbiota of Aedes aegypti. FEMS Microbiol. Ecol.95(1). 10.1093/femsec/fiy 213 (2019).10.1093/femsec/fiy 21330357406 · doi ↗ · pubmed ↗
- 3Romoli, O. & Gendrin, M. The tripartite interactions between the mosquito, its microbiota and plasmodium. Parasit. Vectors. 11(1). 10.1186/s 13071-018-2784-x (2018).10.1186/s 13071-018-2784-x PMC 586161729558973 · doi ↗ · pubmed ↗
- 4Martínez-de la Puente, J. et al. Effects of mosquito microbiota on the survival cost and development success of avian Plasmodium. Front. Microbiol.1110.3389/fmicb.2020.562220 (2021).10.3389/fmicb.2020.562220 PMC 783843933519724 · doi ↗ · pubmed ↗
- 5Zélé, F. et al. Wolbachia increases susceptibility to Plasmodium infection in a natural system. Proc. Biol. Sci.281(1779), 20132837. 10.1098/rspb.2013.2837 (2014).10.1098/rspb.2013.2837 PMC 392407724500167 · doi ↗ · pubmed ↗
- 6Assentato, L. et al. The type of environment has a greater impact on the larval microbiota of Anopheles arabiensis than on the microbiota of their breeding water. FEMS Microbiol. Ecol.101(1). 10.1093/femsec/fiae 161 (2025).10.1093/femsec/fiae 161PMC 1173731839694819 · doi ↗ · pubmed ↗
- 7Mandal, R. K. et al. Gut Bacteroides act in a microbial consortium to cause susceptibility to severe malaria. Nat. Commun.14(1). 10.1038/s 41467-023-42235-0 (2023).10.1038/s 41467-023-42235-0PMC 1057589837833304 · doi ↗ · pubmed ↗
- 8García-Ruiz, O. et al. Sugar diet affects Culex pipiens early-life mortality, biochemical parameters, and immunocompetence. Ecosphere 16(4). 10.1002/ecs 2.70158 (2025).