Novel Genetic Variation in the KIF1A Gene Associated With Cerebellar Vermis Hypoplasia: A Case Report
Gabriela L Lacourt Sosa, Daniela P Torres Rodríguez, Dessire Vergés Santiago, Simon Carlo

TL;DR
A two-year-old girl with developmental delays and cerebellar vermis hypoplasia was found to have a new KIF1A gene mutation, highlighting the need for more research on this rare disorder.
Contribution
A novel de novo KIF1A gene mutation (c.464G>C, p.R155P) is reported in a patient with cerebellar vermis hypoplasia and neurological symptoms.
Findings
The patient had a de novo heterozygous missense mutation in the KIF1A gene at 2q37.3.
MRI findings and symptoms suggest a genetic basis for cerebellar vermis hypoplasia.
The mutation is classified as a variant of uncertain significance but correlates with severe symptoms.
Abstract
KIF1A-associated neuronal disorder (KAND) is a spectrum of disease caused by a genetic mutation in the KIF1A gene. Genetic variants in this gene have been associated with cerebellar vermis hypoplasia, as well as mild to severe symptoms, including ataxia, motor and global developmental delay, and intention tremors. Given the limited number of reported cases, ample research is necessary to effectively manage this condition. A two-year-old girl presents with global developmental, motor, and speech delay, as well as jerky movements, possible athetosis, hand tremors, ataxia, vertical nystagmus, and muscle spasticity in lower limbs. She was referred to the clinic by her neurologist due to abnormal findings in her MRI, which showed cerebellar vermis hypoplasia. These results, together with her symptoms, suggested a genetic basis, for which a chromosomal microarray (CMA) was indicated. Results…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
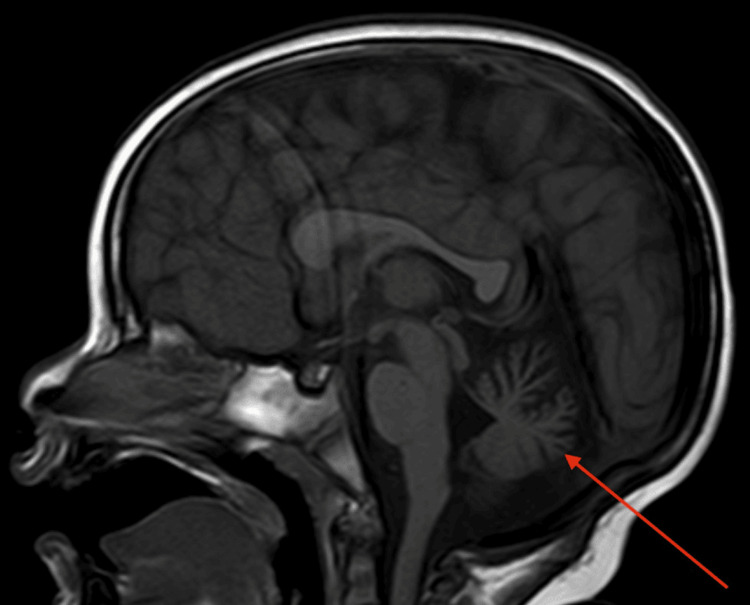 Figure 1
Figure 1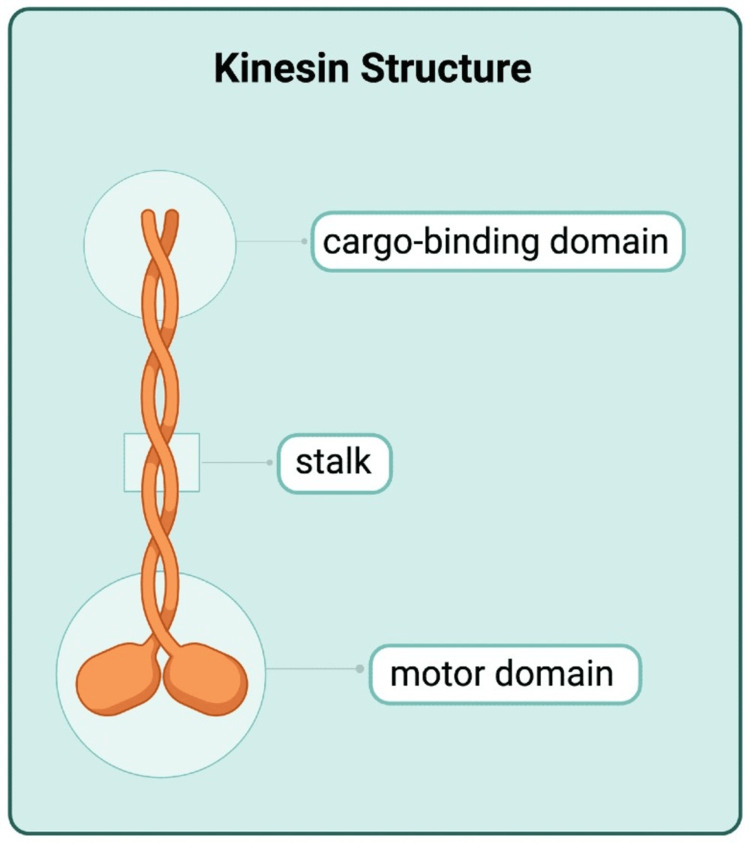 Figure 2
Figure 2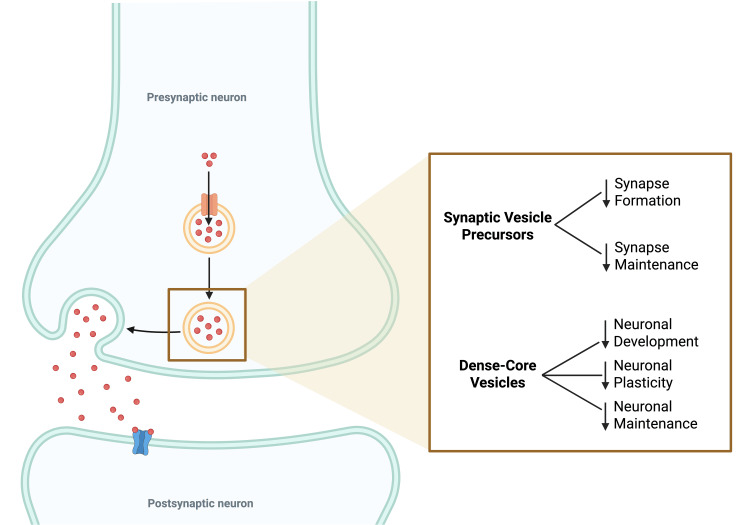 Figure 3
Figure 3| Wild Type | Case Variant | |
| Trait | Arg (R) | Pro (P) |
| Amino Acid Name | Arginine | Proline |
| Polarity/Charge | + Charge | Nonpolar |
| pH | Basic | Neutral |
| Residue Weight | 156 | 97 |
| Hydrophobicity Score | -4.5 | -1.6 |
| Hydrophilicity Score | 3 | 0 |
| Secondary Structure Propensity | Alpha Indifferent/Beta Indifferent | Strong Alpha Breaker/Strong Beta Breaker |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFetal and Pediatric Neurological Disorders · Cancer-related molecular mechanisms research · RNA regulation and disease
Introduction
KIF1A-associated neuronal disorder (KAND) is a genetic disease that causes different degrees of neurological impairment with possible progressive neurodegeneration and neuropathic complications. With a prevalence of 1-10/100,000, its clinical manifestations are generally known to affect multiple organ systems, but their severity depends on the type of genetic mutation in each patient. Some symptoms include, but are not limited to, cerebellar atrophy, cognitive and visual impairment, spastic paraplegia, epilepsy, and peripheral neuropathy [1].
KAND occurs from hereditary or spontaneous variations in the KIF1A gene, which is located on chromosome 2q37.3 and encodes kinesin-like protein KIF1A. This protein is a member of the kinesin family of adenosine triphosphate (ATP)-dependent motor proteins, which are fundamental for the proper anterograde transport of presynaptic and dense-core vesicles, membranous organelles, mRNA, and protein complexes along axonal microtubules. These components that are synthesized in the cell body must be transported to the neuronal synapse for a wide range of essential functions, such as cell survival and development. In addition to cell maintenance and conservation, KIF1A is also responsible for cognitive processes of the brain, such as memory and learning [2].
To understand how different mutations in this gene contribute to the physiological effects seen in mammals, it is essential to first understand the molecular structure and regulation mechanisms of kinesins. Kinesins consist of dimers that have a conserved motor domain, a stalk, and a cargo-binding domain. The motor domain attaches the axonal microtubule and hydrolyzes the ATP needed for movement, while the cargo-binding domain attaches the cargo to be transported. This interaction, together with its proposed auto-inhibitory mechanism, directs its movement toward nerve endings, using the microtubules found on axons as rails so that the cargo to be transported can be moved from the cell body to the neuron terminal [3].
The process of auto-inhibition and activation of KIF1A proteins is a tightly regulated one, with two models of activation having been proposed. The first model suggests a mechanism of auto-inhibition controlled primarily by cargo binding or molecular phosphorylation. It proposes that kinesins that lack cargo are auto-inhibited by existing in a folded state where the tail is free to interact with the motor domain, preventing ATP hydrolysis and, thus, active movement. The second model proposes a conformational change from an inactive monomeric state to an active dimeric state. This change is thought to be induced exclusively by cargo binding to the kinesin tail. In this model, the tail portion of the protein is not directly interacting with the motor domain and inhibiting ATP hydrolysis. This allows it to be readily available to bind the intended cargo and initiate the process of dimerization that allows movement along the microtubules to begin. Previous research using rat models to test the auto-inhibition mechanisms of KIF1A proteins shows that the protein already exists in a dimer form when it is auto-inhibited, supporting the model of cargo-induced activation without the need for conformational changes [4].
After understanding how kinesins are able to transport essential molecules through axonal microtubules, special attention must be paid to the different mutations that have been examined because their pathological manifestation depends on the specific type of mutation in the KIF1A gene. Previous studies have shown that the degree of severity in clinical symptoms depends greatly on whether the genetic mutation is a gain-of-function (GoF) or a loss-of-function (LoF) mutation, as well as the specific location of the mutation in the gene. GoF mutations have been shown to cause the hyperactivation of the protein, leading to excessive motility and synaptic vesicle accumulation at the nerve terminal. On the contrary, LoF mutations reduced the motility of the protein, leading to the accumulation of synaptic vesicles in the cell bodies, rather than at the terminals. These studies associate LoF mutations with more severe phenotypes that include hereditary spastic paraplegia and intellectual disability, while GoF mutations show milder phenotypes with hereditary spastic paraplegia but no intellectual disability [4].
We present a case of a patient with a variant of uncertain significance in the KIF1A gene showing symptoms of developmental delay with visual disturbances, as well as neuromuscular damage.
Case presentation
A two-year-old Puerto Rican girl presented with abnormal movements and developmental delay. She was evaluated by a neurologist who made an initial diagnosis of global developmental delay and gross motor delay, as well as language delay. A previous brain MRI showed cerebellar vermis hypoplasia (Figure 1).
Brain MRI scan of the patient.Red arrow: the cerebellum showing vermis hypoplasia.
Upon further evaluation, she showed the hypertonia of bilateral lower extremities and bilateral upgoing toes on plantar reflex, which were indicative of a positive Babinski sign (Figure 1). Additionally, she demonstrated vertical nystagmus, head oscillations, and hand tremors when initiating movement to grab objects. These symptoms directed the diagnosis toward an upper motor neuron lesion, as well as a cerebellum lesion. Subsequently, a diagnosis of cerebral palsy was suggested. The final diagnoses by her neurologist were other disorders of psychological development, specific disorder of motor function, mixed-receptive-expressive language disorder, unspecified tremor, unspecified nystagmus, unspecified abnormal involuntary movements, and transient alteration of awareness. She was referred to a geneticist for the evaluation of possible causes of the disease.
Upon evaluation, additional symptoms of jerky movements, possible athetosis, ataxia, muscle spasticity in lower limbs, and recurrent infections were noted. Due to this clinical presentation and the absence of abnormal metabolic panels or any contributing family history, a chromosomal microarray (CMA) was ordered.
Results for this test showed two regions of allelic homozygosity (ROH) with a combined length of approximately 16 Mb, including regions of chromosomes 1 and 7. Next, a whole-exome sequencing was ordered, which reported a variant of uncertain clinical significance on gene KIF1A, specifically c.464G>C (p.R155P), causing the arginine at amino acid position 155 to be replaced by a proline. This variation was described as autosomal dominant, heterozygous, and related to KANDs.
Discussion
The severity of KAND’s pathological phenotype lies in the fact that adequate neurological function depends on this tightly regulated system of transport and communication between axons, which, in turn, has been shown to largely depend on kinesin proteins with KIF1A playing a central role (Figure 2).
Schematic representation of the KIF1A protein structure.Created in BioRender by Lacourt Sosa GL (2025) (https://BioRender.com/kljj52z).
The specific degree of the disease’s pathogenicity has been shown to be determined by the type of mutation, with previous case studies showing variable phenotypes that correlate with different mutations in the same gene. However, it is believed that mutations affecting the motor domain of the KIF1A protein are implicated in the highest level of clinical manifestations, whereas mutations affecting other regions of the protein have been shown to be nonpathogenic [5].
Previous research regarding the effect of KIF1A defects on the cellular mechanism of individuals reveals how different errors in microtubular transport lead to different cellular phenotypes. This variation in clinical manifestations occurs due to the importance and specificity of each cargo transported by the kinesin (Figure 3).
Schematic representation of the neuronal synapse and the effect of transport components on neurological status.Created in BioRender by Lacourt Sosa GL (2025) (https://BioRender.com/qzjw2on).
The defective transport and localization of synaptic vesicle precursors have been shown to decrease synapse formation and maintenance, while defective dense-core vesicles affect neuronal development, maintenance, and plasticity. Consequently, patients with genetic variants in the KIF1A gene can show varying degrees of intellectual disability, as well as neurodegeneration. Another important substance that is affected in genetic mutations of this sort is brain-derived neurotrophic factor. This neurotrophin has been known to be expressed in critical areas of the brain, such as the hippocampus, cortex, pyramidal tracts, and cerebellum, which are thought to be involved in hereditary spastic paraplegia-like disorders. In addition to brain-derived neurotrophic factor, other substances that are essential for neurological function are vesicular monoamine transporter 2 (VME2), netrin-1, and autophagy-related protein 9 (ARP9). VME2 is a protein transported via dense-core vesicles that is involved in the transport and accumulation of monoamines into synaptic vesicles, with norepinephrine, dopamine, and serotonin being the main ones affected. On the other hand, netrin-1 is a protein involved in cortical and cerebellar migration, while ARP9 is responsible for protein clearance and damaged organelle recycling. Previous research has shown that ARP9 knockout mice have an accelerated rate of neurodegeneration, confirming the chemical’s vital function [6]. This could directly correlate with the phenotype of our subject, which shows signs of muscle spasticity and cerebellar atrophy that could potentially be caused by the improper transport of these chemicals.
The whole exome sequencing report performed on our subject revealed a unique genetic variant in the KIF1A gene that had not been previously reported. The KIF1A c.464G.C genetic mutation located in amino acid 155 alters the molecular properties of the protein in a great manner, affecting properties such as hydrophobicity, polarity, and pH, as well as residue weight (Table 1).
Genetic analysis predicts it to be deleterious by in silico analysis, as the amino acid change region happens to be in an area with high missense constraint. This means that the location affected in the gene has been highly conserved in available vertebrate species due to probable low tolerance to changes that lead to highly disrupted proteins. These defective proteins are not compatible with evolutionary pressures, as they confer a survival disadvantage to the organism bearing the genetic mutation. Deleterious mutations of this kind have been shown to be removed from the genetic pool through the process of negative selection, making them extremely rare but also severely pathogenic. Although these analyses suggest a deleterious effect of the variant, future studies including segregation analyses, functional assays, and advanced computational modeling would aid to officially classify the variant as pathogenic under American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines.
In addition to the mutation in the KIF1A gene that is correlated to KAND, the CMA performed on our subject revealed a region of allelic homozygosity in chromosomes 1 and 7, which could be indicative of familial consanguinity [7]. The genetic analysis states that even though this result is not sufficient for a definitive diagnosis of any specific condition, it does raise the risk of a recessive Mendelian disorder with a causative genetic variant in this specific region. This finding is worth analyzing in conjunction with the variant in the KIF1A gene as a possible cause for the subject’s pathogenic condition, as no additional analysis was conducted to identify potentially pathogenic recessive variants within these ROHs.
Conclusions
The rarity of the genetic mutation, as well as the distinct clinical presentation of the subject in this case report, exemplifies the need for more extensive research regarding the specific mechanism of KANDs. More information is needed to identify whether the damage caused in the kinesin-like KIF1A protein is due to defects in its ATP hydrolysis ability, protein structure, or its cargo-binding abilities. There is optimism that ascertaining the specific defect in each individual patient’s KIF1A protein product will help tailor a more assertive and effective line of treatment for KAND.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Care recommendations 7 2024 https://www.kif 1a.org/kand/kif 1a-associated-neurological-disorder/
- 2KIF 1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons Neurosci Lett Lo KY Kuzmin A Unger SM Petersen JD Silverman MA 1681734912011 https://www.sciencedirect.com/science/article/abs/pii/S 030439401100022 X 2125692410.1016/j.neulet.2011.01.018 · doi ↗ · pubmed ↗
- 3Insight into the regulation of axonal transport from the study of KIF 1A-associated neurological disorder J Cell Sci Chiba K Kita T Anazawa Y Niwa S 01362023 https://journals.biologists.com/jcs/article/136/5/jcs 260742/286709/Insight-into-the-regulation-of-axonal-transport 10.1242/jcs.26074236655764 · doi ↗ · pubmed ↗
- 4Mammalian kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition P Lo S Biol Hammond JW Cai D Blasius TL 072009 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC 2661964/10.1371/journal.pbio.1000072 PMC 266196419338388 · doi ↗ · pubmed ↗
- 5A de novo low-frequency mosaic variant of KIF 1A causes hereditary spastic paraplegia: a literature review Ann Hum Genet Liu M Li B Wang X 158165872023 https://onlinelibrary.wiley.com/doi/10.1111/ahg.125033689678410.1111/ahg.12503 · doi ↗ · pubmed ↗
- 6Going too far is the same as falling short†: kinesin-3 family members in hereditary spastic paraplegia Front Cell Neurosci Gabrych DR Lau VZ Niwa S Silverman MA 419132019 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC 6775250/3161625310.3389/fncel.2019.00419 PMC 6775250 · doi ↗ · pubmed ↗
- 7Interpretation and reporting of large regions of homozygosity and suspected consanguinity/uniparental disomy, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG)Genet Med Gonzales PR Andersen EF Brown TR 255261242022 https://pubmed.ncbi.nlm.nih.gov/34906464/3490646410.1016/j.gim.2021.10.004 · doi ↗ · pubmed ↗