De novo missense mutation in MYT1l leading to autosomal dominant intellectual disability 39 and autism spectrum disorder: a case report
Xin Wang, Shuangzhu Lin, Yang Chen, Yangfan Qi, Xiaoyu Sun, Wanqi Wang, Kai Jiang

TL;DR
A young girl with developmental delays and autism was found to have a new mutation in the MYT1l gene, which is linked to a rare genetic disorder.
Contribution
This case report confirms the pathogenicity of a previously uncharacterized MYT1l variant and expands the known phenotype of MRD39.
Findings
A de novo missense variant in MYT1l (c.1695G > T; p.Arg565Ser) was identified in a patient with global developmental delay and autism.
The variant was classified as Likely Pathogenic based on ACMG criteria PM6 and PM2.
The case expands the phenotypic spectrum of MYT1l-related neurodevelopmental disorders.
Abstract
Autosomal dominant intellectual disability type 39 (MRD39; OMIM # 616521) is caused by heterozygous mutation in the MYT1l gene on chromosome 2p25.3. The MYTL1 encoded protein belongs to a novel class of cystein-cystein-histidine-cystein zinc finger proteins that function in the developing mammalian central nervous system. We report a 1-year-6-month-old girl presenting with global developmental delay (GDD) and autistic behaviors, demonstrating inability to stand independently, crawling mobility, poor response to name calling, and impaired joint attention. Initial developmental assessments yielded a Griffiths Mental Development Scale score of 57 and an ADOS-2 score of 11. Following 20 months of systematic rehabilitative training, the patient achieved independent ambulation, could follow simple commands, and produced phrases under 10 words, though suboptimal response to name calling and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
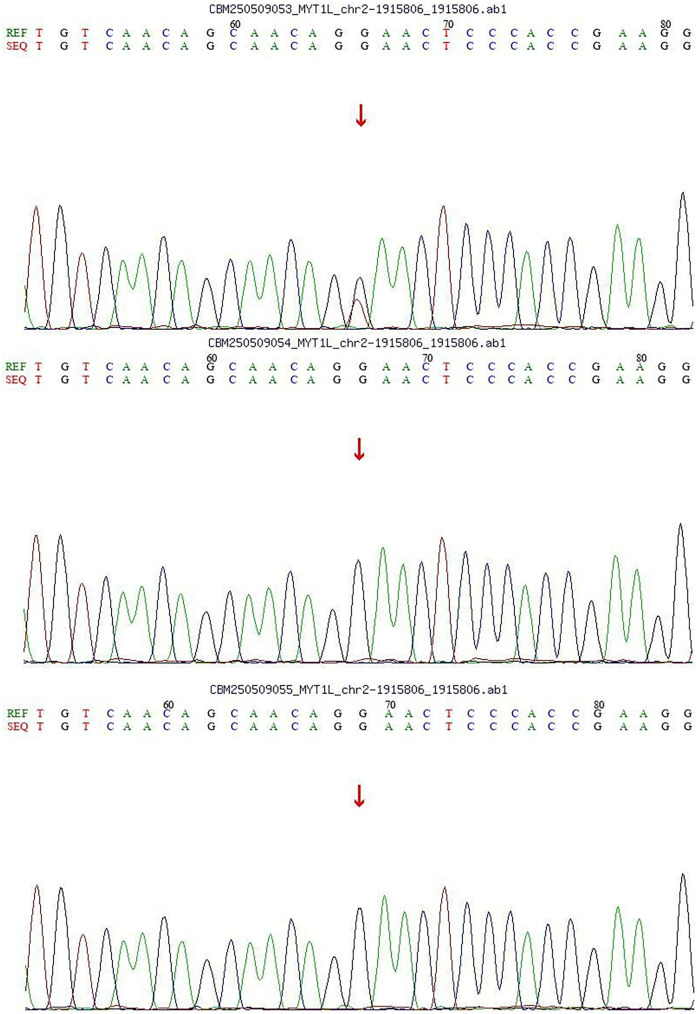 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetics and Neurodevelopmental Disorders · Genomics and Rare Diseases · Genomic variations and chromosomal abnormalities
Introduction
1
Autosomal dominant intellectual disability type 39 (MRD5; OMIM #616521) is caused by heterozygous mutation in the MYT1l gene on chromosome 2p25.3 (1). The MYT1l gene comprises 12 exons and encodes a protein of 1,230 amino acids (1, 2). The core clinical manifestations of MRD39 include intellectual disability, delayed language development, autism spectrum disorder, with partial patients exhibiting distinctive facial features and epileptic seizures (3–5).
In this study, we present a case of a patient diagnosed with MRD39, characterized by significant developmental delay and autistic features, yet notably lacking seizures. Whole-exome sequencing (WES) revealed a de novo heterozygous missense mutation in the MYT1l gene, which aligns with the patient's clinical phenotype. This finding contributes to the expanding knowledge of MYT1l variants and their associated clinical presentations.
Case presentation
2
Chief complaint
2.1
A 1-year-6-month-old girl was referred for evaluation of developmental delay observed over the preceding 8 months.
The patient presented with inability to ambulate independently since birth. This female infant was born at 37 weeks' gestation (G2P2) via spontaneous vaginal delivery, with a birth weight of 3.0 kg. There was no history of intrauterine hypoxia or neonatal resuscitation. She was formula-fed postnatally. Developmental milestones included: head control at 4 months, rolling over at 6 months, independent sitting at 11 months, stable sitting without support at 14 months, crawling at 15 months, and standing independently at 16 months. Clinical observations revealed poor response to name calling, limited eye contact, absence of stranger anxiety, and prominent repetitive head-banging behavior. Initial developmental assessments yielded a Griffiths Mental Development Scale score of 57 and an ADOS-2 score of 11.
Physical examination
2.2
Weight 10 kg, head circumference 46 cm. The child was alert and responsive with regular respirations. No hypopigmented patches or café-au-lait spots were observed. The anterior fontanelle was closed. Ocular motility was normal, and the pupils were isocoric with normal light reflexes. No nuchal rigidity was noted. Cardiopulmonary and abdominal examinations were unremarkable. Cranial nerve assessment showed no abnormalities. Muscle strength and tone were normal in all extremities. Knee jerks and abdominal reflexes were present. Ankle clonus was absent. Kernig's sign, Brudzinski's sign, Babinski sign, and Oppenheim sign were negative bilaterally.
Family history
2.3
The patient's father and mother are healthy, and the patient's 8-year-old brother is healthy.
Routine clinical examinations
2.4
A comprehensive evaluation, including complete blood count, urinalysis, stool analysis, assessments of liver and kidney function, cardiac enzyme levels, electrolyte balance, thyroid function, and both blood and urine genetic metabolic screenings, indicated no abnormalities. Electroencephalography (EEG) revealed no abnormalities, and brain magnetic resonance imaging (MRI) showed no structural lesions.
Genetic analysis
2.5
With the consent of the parents, we performed whole exome sequencing (WES) to identify potential pathogenic variants.Following sequencing, a stepwise filtering approach was applied to prioritize candidates: common polymorphisms (allele frequency >0.1% in aggregate population databases including gnomAD, 1,000 Genomes, and ExAC) were excluded, and focus was placed on rare, protein-altering variants. Given the patient's phenotype and negative family history, we prioritized de novo variants. Whole-exome sequencing (WES) revealed a de novo heterozygous missense variant in the MYT1l gene [c.1695G > T; p.(Arg565Ser)]. Parental Sanger sequencing confirmed that both parents carried the wild-type allele for this variant (Figure 1). Based on the ACMG/AMP guidelines, the variant was assessed as Likely Pathogenic, supported by evidence PM6 (confirmed de novo status) and PM2 (absence in population databases).
Sanger diagram.
Final diagnosis
2.6
MRD39, ASD.
Treatment
2.7
A comprehensive rehabilitation program spanning 20 months was implemented for the patients.
Outcome and follow-up
2.8
Following 20 months of systematic rehabilitation training, the patient achieved independent walking and short-distance running, and can speak short phrases of fewer than 10 words. However, persistent deficits include reduced eye contact, poor response to name calling, and stereotypical head-banging behavior. Re-evaluation showed a Griffiths score of 59 and ADOS-2 score of 10.
Discussion
3
The MYT1l gene is integral to neuronal differentiation and neural development. Studies demonstrate that MYT1l sustains neuronal identity by repressing the expression of genes associated with non-neuronal lineages. This repression occurs through its interaction with the Sin3b complex, which is recruited via the N-terminal domain of MYT1l (6). Furthermore, MYT1l exhibits similar genomic binding sites in both neurons and fibroblasts and is typically found in an open chromatin configuration, emphasizing its vital role in maintaining neuronal identity (6).
Beyond its function in neurons, MYT1l also facilitates the differentiation of oligodendrocyte precursor cells (OPCs). Evidence indicates that MYT1l is expressed during myelin formation and remyelination, with its expression being regulated by binding to the Olig1 promoter, thereby promoting OPC differentiation (7). These biological functions position MYT1l as a promising therapeutic target for demyelinating diseases.
In neurodevelopmental disorders, loss-of-function mutations in the MYT1l gene are correlated with intellectual disability, autism spectrum disorder, and obesity (2, 8). Studies suggest that MYT1l deficiency results in the aberrant activation of neuronal developmental programs, consequently impairing neuronal maturation and function[ (9). Moreover, the absence of MYT1l leads to an imbalance in neuronal proportions, predominantly affecting neuronal maturation processes. This developmental imbalance persists throughout the developmental timeline (10).
Mutations in the MYT1l gene are also implicated in schizophrenia and other neuropsychiatric disorders. Research has demonstrated that microdeletions in MYT1l are significantly associated with childhood-onset schizophrenia, indicating that MYT1l may contribute to the pathogenesis of these conditions (11). Additionally, MYT1l expression is linked to memory-related processes in the brain, influencing these processes by regulating the proliferation/differentiation switch of ID-bHLH factors (12).
In this case, both parents were healthy at the child's birth, with no history of intrauterine asphyxia or amniotic fluid contamination, and no family history of genetic disorders. Postnatally, the patient exhibited global developmental delay accompanied by manifestations of autism spectrum disorder. Based on the clinical presentation, whole-exome sequencing (WES) identified a de novo heterozygous missense variant in the MYT1l gene [c.1695G > T; p. (Arg565Ser)], which was confirmed by Sanger sequencing to segregate within the family according to genetic co-segregation principles. According to the ACMG/AMP guidelines, this variant was classified as “Likely Pathogenic” based on criteria PM6 (confirmed de novo status) and PM2 (absence in population databases). Despite the variant's classification as “Likely Pathogenic” and the need for functional validation, the significant overlap between the patient's phenotype and the established MYT1l-associated neurodevelopmental profile, in the absence of other plausible genetic findings, strongly implicates this variant as the most likely underlying cause. Therefore, integrating the compelling clinical and genetic evidence, a definitive diagnosis of Autosomal Dominant Intellectual disability 39 and Autism Spectrum Disorder was established.
The MYT1l variant at position 565 (p.Arg565Ser) (ID: 801647) lacks a definitive pathogenicity assignment in the ClinVar database, necessitating its evaluation using the PM6 and PM2 criteria. While our classification of “Likely Pathogenic” is robustly supported by de novo status and population frequency data, the application of other ACMG criteria warrants further discussion. the PS1 criterion (identical amino acid change as a known Pathogenic variant) could not be applied because this variant lacks independent basis for a “Pathogenic” or “Likely Pathogenic” designation. Similarly, the PP5 criterion (reported as Pathogenic by an authoritative source but with unpublished data) was also not met. Compared to the well-established loss-of-function mechanisms in MYT1l, the pathogenicity of this missense variant remains to be fully elucidated through functional studies. our case, combined with existing ClinVar submissions, adds to the body of evidence linking this specific variant to neurodevelopmental disorders and highlights the need for further evidence to strengthen its classification criteria.
After 20 months of structured rehabilitation training, the patient exhibited notable advancements in motor skills, including the ability to walk independently and run short distances, as well as enhancements in language abilities, demonstrated by the production of short phrases consisting of fewer than 10 words. Nevertheless, the patient continued to experience persistent deficits, such as diminished eye contact, inadequate response to auditory cues such as name calling, and repetitive head-banging behavior. Remarkably, in contrast to previously documented cases, this patient presented with normal findings on brain MRI, EEG results, and no history of seizure episodes.
We report a case of MYT1l-related MRD39, distinguished by pronounced global developmental delay and autism spectrum disorder, and characterized by a novel c.1695G > T missense variant that has not been previously documented. This finding broadens the current understanding of the mutation spectrum and phenotypic variability associated with MYT1l, thereby advancing insights into the genotype-phenotype correlations in conditions related to MYT1l.
Patient perspective
The patient's legal guardian provided written informed consent for the publication of this case report.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1de Ligt J Willemsen M Hvan Bon BW Kleefstra T Yntema HG Kroes T Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. (2012) 367(20):1921–9. 10.1056/NEJ Moa 120652423033978 · doi ↗ · pubmed ↗
- 2Mayo S RosellóM Monfort S Oltra S Orellana C Martínez F. Haploinsufficiency of the MYT 1l gene causes intellectual disability frequently associated with behavioral disorder. Genet Med. (2015) 17(8):683–4. 10.1038/gim.2015.8626240977 · doi ↗ · pubmed ↗
- 3Boeri S Scala M Madia F Perucco F Vozzi D Capra V MYT 1l Variant inherited by a mosaic father in a case of severe developmental and epileptic encephalopathy. Epileptic Disord. (2023) 25(6):874–9. 10.1002/epd 2.2014137518898 · doi ↗ · pubmed ↗
- 4De Rocker N Vergult S Koolen D Jacobs E Hoischen A Zeesman S Refinement of the critical 2p 25.3 deletion region: the role of MYT 1l in intellectual disability and obesity. Genet Med. (2015) 17(6):460–6. 10.1038/gim.2014.12425232846 · doi ↗ · pubmed ↗
- 5Blanchet P Bebin M Bruet S Cooper GM Thompson ML Duban-Bedu B MYT 1l Mutations cause intellectual disability and variable obesity by dysregulating gene expression and development of the neuroendocrine hypothalamus. P Lo S Genet. (2017) 13(8):e 1006957. 10.1371/journal.pgen.100695728859103 PMC 5597252 · doi ↗ · pubmed ↗
- 6Mall M Kareta MS Chanda S Ahlenius H Perotti N Zhou B Myt 1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature. (2017) 544(7649):245–9. 10.1038/nature 2172228379941 PMC 11348803 · doi ↗ · pubmed ↗
- 7Shi Y Shao Q Li Z Gonzalez GA Lu F Wang D Myt 1l promotes differentiation of oligodendrocyte precursor cells and is necessary for remyelination after lysolecithin-induced demyelination. Neurosci Bull. (2018) 34(2):247–60. 10.1007/s 12264-018-0207-929397565 PMC 5856724 · doi ↗ · pubmed ↗
- 8Stevens S Jvan Ravenswaaij-Arts CM Janssen JW Klein Wassink-Ruiter J Svan Essen AJ Dijkhuizen T MYT 1l Is a candidate gene for intellectual disability in patients with 2p 25.3 (2pter) deletions. Am J Med Genet A. (2011) 155(11):2739–45. 10.1002/ajmg.a.3427421990140 · doi ↗ · pubmed ↗