Design, synthesis, and biological evaluation of phenylisoxazole-based histone deacetylase inhibitors
Xiaofei Qin, Meng Han, Peng Hu, Huadong Que, Zhenlei Shao, Dong Yan, Afzal Shaik, Afzal Shaik, Afzal Shaik

TL;DR
This study designs and tests new HDAC1 inhibitors that show strong anti-cancer activity in prostate cancer cells.
Contribution
A new series of phenylisoxazole-based HDAC1 inhibitors with potent anti-proliferative activity is developed.
Findings
Compound 17 showed 86.78% HDAC1 inhibition at 1000 nM.
Compound 17 had an IC50 of 5.82 μM against prostate cancer PC3 cells.
Compound 17 showed no significant toxicity to normal prostate cells.
Abstract
Histone deacetylases (HDACs) mediate the removal of acetyl groups from lysine residues in both histone and non-histone proteins, and have been regarded as promising targets for drug discovery. As a central member of HDAC family, HDAC1 has been found to be closely linked to the occurrence and development of prostate cancer. In this study, we designed and synthesized a new series of 3-phenylisoxazole HDAC1 inhibitors based on the hit 7, identified by in-house compound library screening. The structure-activity relationship studies (SARs) indicated that the R1 position was relatively tolerated for activity. The linker length at R2 exerted a significant influence on activity with the relative orders of butyl > propyl > ethyl > methyl. Among synthetic 16 compounds, compound 17 indicated the strongest HDAC1 inhibitory effect with the inhibition rate of 86.78% at the concentration of 1000 nM.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
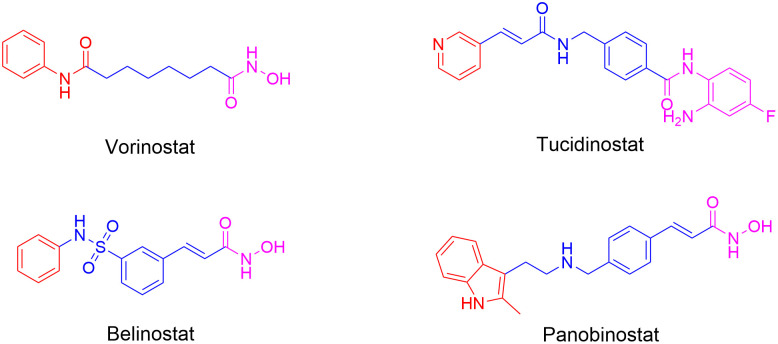 Fig 1
Fig 1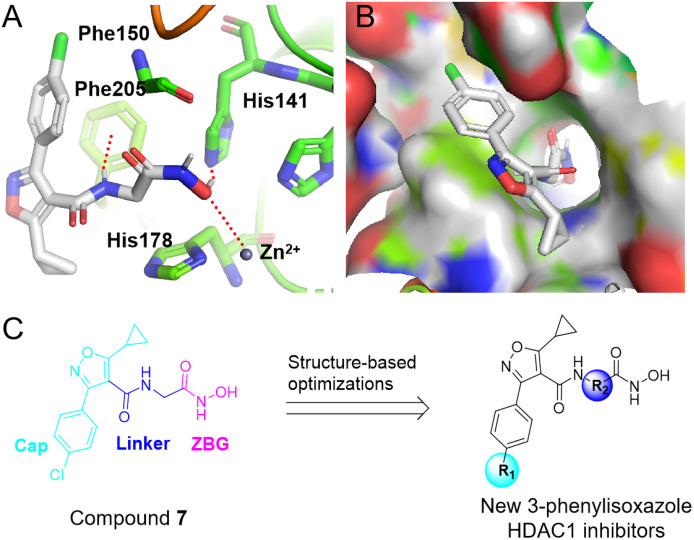 Fig 2
Fig 2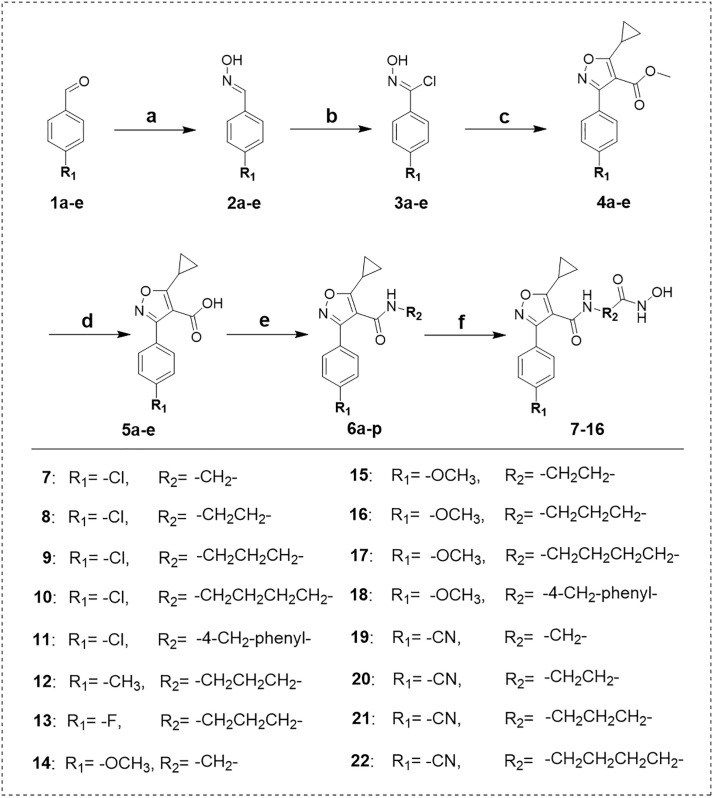 Fig 3
Fig 3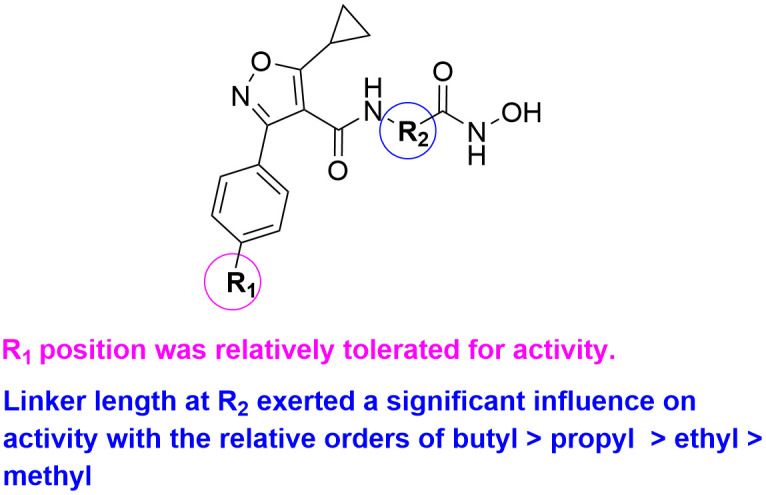 Fig 4
Fig 4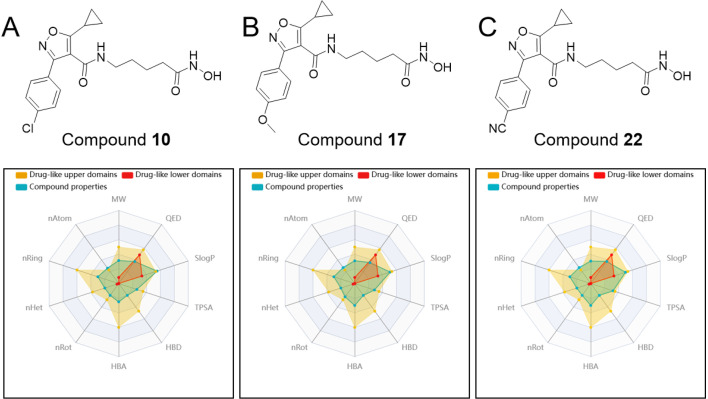 Fig 5
Fig 5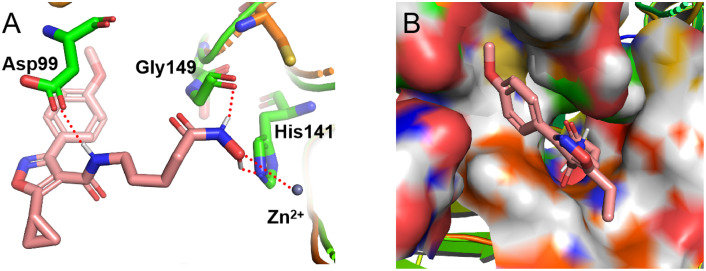 Fig 6
Fig 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHistone Deacetylase Inhibitors Research · Epigenetics and DNA Methylation · Prostate Cancer Treatment and Research
1. Introduction
Histone modification serves as a pivotal epigenetic mechanism, particularly manifesting through the acetylation-deacetylation dynamics governed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDACs predominantly mediate the removal of acetyl groups from lysine residues in both histone and non-histone proteins, thereby inducing chromatin remodeling and orchestrating the transcriptional modulation of critical apoptotic and cell cycle regulatory genes [1–3]. Eleven zinc-dependent HDAC isoforms have been found in humans, phylogenetically divided into four classes based on structural domains, localization patterns, and homology. These classes consist of Class I (HDAC1/2/3/8), Class IIa (HDAC4/5/7/9), Class IIb (HDAC6/10), and Class IV (HDAC11) [4,5]. HDAC1 is usually checked in different organs. During the mitotic phase, HDAC1 protein is critical for condensing of chromatin, separating of chromosomes, and formatting of spindles [6]. Additionally, HDAC1 participates in multiple biological processes, such as red blood cell production, liver regeneration, programmed cell death, formation of new blood vessels, and cell division regulation [6]. HDACs establish dysregulation in many human diseases and are recognized as important therapeutic targets for cancers [3,7], diabetes [8], inflammatory processes [9,10], and cardiac diseases [11,12] and so on. In particular, the overexpression of HDAC1 plays a significant role in the progression of prostate cancer and is linked to a poor prognosis [13,14]. The expression level of HDAC1 is positively correlated with the abnormal proliferation of prostate cancer PC3 cells, which could be significantly reversed on treatment with an HDAC inhibitor TSA [15,16]. Moreover, HDAC1 downregulation results in E-cadherin expression and following inhibition of cell motility and invasion [17]. Targeting HDAC1 has been regarded as a promising approach for the treatment of prostate cancer.
Recently, the development of HDAC inhibitors (HDACIs) has emerged as a new strategy for innovative drug discovery. HDACIs design typically follows a pharmacophore model composed of three core elements: (ⅰ) a cap group that interacts with the entrance of catalytic center, (ⅱ) a linker domain linking the cap to zinc-binding group (ZBG), and (ⅲ) the ZBG moiety itself [18]. To date, five HDAC inhibitors (HDACIs), including Vorinostat (SAHA), Romidepsin, Tucidinostat, Panobinostat, and Belinostat, have been approved for use in hematologic cancers, while many clinical trials are currently assessing their effects in solid tumors (Fig 1) [18,19]. However, significant adverse effects associated with these approved HDACIs have been gradually discovered, including myelosuppression, gastrointestinal, cardiotoxicity, and hepatic abnormalities etc [20,21]. Therefore, the discovery of new HDAC inhibitors with low toxicity still holds significant importance and value.
The structures of representative HDAC inhibitors.
We previously carried out an in-house compound library screening, providing a 3-phenylisoxazole derivative 7, which showed 9.30% inhibitory activity against HDAC1 at 1000 nM (Fig 2C). Based on compound 7, we herein performed further structural optimizations and structure-activity relationship studies, yielding a new series of 3-phenylisoxazole analogues, of which molecule 17 showed strong HDAC1 inhibitory activity, potent antiproliferative activity, good drug-like properties and low toxicity. Overall, this study offered a new structural skeleton for the development of drugs targeting HDACs.
The design strategy of new 3-phenylisoxazole HDAC1 inhibitors.(A-B) The predicted binding modes of hit 7 (white) with HDAC1 (PDB: 5ICN). (C) The design scheme of new 3-phenylisoxazole HDAC1 inhibitors.
2. Results and discussion
2.1. Structure-based drug design of new 3-phenylisoxazole HDAC inhibitors
To discover new HDAC inhibitors, we conducted an in-house compound library screening, offering hit 7, which indicated 9.3% inhibitory effect against HDAC1 at 1000 nM (Fig 2C). The molecular docking study shows that the amino hydrogen of hit 7 forms a π-H interaction with the benzene ring of Phe205. The hydroxyl hydrogen in the compound 7 has a hydrogen bond interaction with His141. In addition, the carbonyl oxygen in compound 7 demonstrates a relatively strong zinc-binding ability. Besides, the 4-chlorobenzyl group sits in a hydrophobic pocket. Based on these binding modes, structural optimizations and SAR studies were performed via focusing on R_1_ and R_2_ position (Fig 2C), in hope of further improving the hydrophobic interactions and zinc-binding ability.
2.2. Chemistry
The overall synthetic routes for new 3-phenylisoxazole derivatives were illustrated in Fig 3. Condensation of commercially available 1a-e with hydroxylamine yielded intermediates 2a-e, which were further chlorinated using N-chlorosuccinimide, resulted in the production of intermediates 3a-e. intermediates 3a-e were used to react with methyl 3-cyclopropyl-3-oxopropionate in the presence of triethylamine, providing cyclization intermediates 4a-e. Hydrolysis of intermediates 4a-e with sodium hydroxide produced key intermediates 5a-e, which were condensed with different substituted amines, providing intermediates 6a-p. Ammonia hydrolysis of intermediates 6a-p with hydroxylamine under sodium hydroxide catalysis generated title compounds 7–16.
The Synthetic route of 3-phenylisoxazole derivatives.Reagents and conditions: (a) Hydroxylamine, EtOH, 60°C, 2 h; (b) N-Chlorosuccinimide, DMF, 40°C, 2 h; (c) Methyl 3-cyclopropyl-3-oxopropionate, triethylamine, EtOH, 0°C-re, 5-6 h; (d) NaOH, H2O, 80°C, re, 1 h; (e) Suitable amine compounds, DIPEA, EDCI, rt, 2 h; (f) Hydroxylamine, NaOH, H2O, rt, 1 h.
2.3. Structure-activity relationship (SAR) studies
The inhibitory effects of compounds against HDAC1 were expressed in terms of inhibition rates at a single concentration. Vorinostat (SAHA), an approved HDAC inhibitor, was served as the positive control [22]. As shown in Table 1, compound 7 tethering a methyl group at R_2_ position suppressed HDAC1 with the inhibition rate of 9.30% at the concentration of 1000 nM. The extension of methyl group in compound 7 with longer linkers resulted in compounds 8–11. We observed that the linker length at R_2_ position exerted a significant influence on activity with the relative orders of butyl group > propyl group > ethyl group > methyl group (10 vs. 9 vs. 8 vs. 7). In particular, molecule 10 indicated the strongest binding affinity on HDAC1 with the inhibition rate of 79.85%, about 6-folds more potent than that of compound 7. Replacement of butyl group in compound 10 with a benzyl group provided derivative 11, which had the inhibition rate of 63.38%, slightly weaker activity in comparison to that of analogue 10. Changing the chlorine atom of compounds 7–11 to methoxy, or cyano group resulted in compounds 14–22, which exhibited similar activity trend. Next, the SARs studies of R_1_ position were further conducted. As demonstrated in Table 1, compound 9 with a chlorine atom at R_1_ position displayed the inhibition rate of 13.43%. However, translating the chlorine atom of compound 9 to other substituents, such as methyl (12), fluorine (13), methoxy (16) or cyano (21), did not resulted in an obvious alteration of HDAC1 inhibitory activity, suggesting that the R_1_ position was relatively tolerated for activity. The SARs summary was depicted in Fig 4.
Table 1: The HDAC1 inhibitory activity of 3-phenylisoxazole derivatives 6-20.
The summary for SAR studies of 3-phenylisoxazole derivatives.
2.4. Drug-likeness studies of 3-phenylisoxazole derivatives 10, 17 and 22
Giving the potent inhibitory effects of compounds 10, 17 and 22 towards HDAC1, we further assessed their drug-like properties through the admet SAR 3.0 website (https://lmmd.ecust.edu.cn/admetsar3/predict_submit.php). As indicated in Table 2, molecule 17 had a molecular weight of 373.41, number of atoms of 27, number of heteroatoms of 8, number of rings of 3, number of rotatable bonds of 9, number of hydrogen bond acceptors of 6, number of hydrogen bond donors of 3, topological polar surface area of 113.69, and the logarithm of n-octanol/water distribution coefficient of 2.63. These properties made compound 17 fully satisfy both lipinski rule and pfizer rule (Table 2), which were generally applied to estimate the physicochemical properties and drug-like property of drugs. Meanwhile, molecules 10 and 22 also displayed similar drug-like properties (Table 2). Besides, compounds 10, 17 and 22 indicated almost no significant effect on hERG, suggesting that these molecules probably did not cardiotoxicity. The analysis presented in Fig 5, generated by the ADMET SAR 3.0 website, further substantiates that these 3-phenylisoxazole derivatives possessed favorable drug-like properties.
Table 2: The predicted drug-like parameters of representative compounds.
The drug-like analysis diagrams of the compounds 10, 17 and 22.
2.5. Anti-proliferative activity of representative 3-phenylisoxazole derivatives
Considering the potential therapeutic efficacy of HDAC1 inhibitors for prostate cancer, PC3 cells were used to evaluate in vitro anticancer activity of representative 3-phenylisoxazole derivatives 10 and 17. The results showed that molecule 10 inhibited PC3 cells with the IC_50_ value of 9.18 μM (Table 3), slightly weaker in comparison to that of compound 17 (IC_50_ = 5.82 μM). Interestingly, both compounds indicated no evident toxicity on prostate normal WPMY-1 cells. These results displayed that both compounds possessed potent anti-prostate cancer activity and low toxicity.
Table 3: The anti-proliferative activity of representative 3-phenylisoxazole derivatives.
2.6. Molecular docking study
Encouraged by the potent activity, weak toxicity, and good drug-like properties of compound 17, we next explored its binding modes with HDAC1 protein. As shown in Figs 2 and 6, compared to compound 7, molecule 17 can better occupy the activity pocket of HDAC1. The amino hydrogen in compound 17 has a hydrogen bond interaction Asp99 and Gly149, respectively. Another hydrogen bond is formed between hydroxyl hydrogen of derivative 17 and imidazole nitrogen atom. Besides, the carbonyl oxygen of compound 17 is observed to have a strong zinc-binding effect. These interactions may be responsible for the high activity on HDAC1 of compound 17.
The binding modes of compound 17 with HDAC1.(A) Interactions between 17 (pink) and residues of HDAC1. (B) The binding pocket surface of HDAC1 and 17 (PDB: 5ICN).
3. Conclusion
In summary, a new class of 3-phenylisoxazole derivatives were designed and synthesized as potent HDAC1 inhibitors. The SARs exhibited that the R_1_ position was well tolerated for activity. The linker length at R_2_ showed a significant influence on activity with the relative orders of butyl > propyl > ethyl > methyl. The representative compound 17 displayed significant inhibitory potency against HDAC1 with the inhibition rate of 86.78% at the concentration of 1000 nM. MTT assay showed that derivative 17 possessed strong cytotoxicity toward prostate cancer PC3 cells, yet had no obvious influence on the growth of normal WPMY-1 cells. The docking study presented that derivative 17 could be well matched with the active pockets of HDAC1. Besides, molecule 17 exhibited good drug-like properties. Overall, phenylisoxazole derivative 17 could serve as a lead compound for further optimizations in the treatment of prostate cancer.
4. Experimental section
4.1. General
Melting points were determined using a WRS-1A digital melting point apparatus. Thin-layer chromatography (TLC) was performed on silica gel-precoated glass plates with visualization under UV light (254 nm or 365 nm). All reagents and solvents were commercially sourced and used as received. Mass spectrometry data were acquired on a Waters Acquity UPLC system operating in positive electrospray ionization (ESI^+^) mode. NMR spectra (¹H: 400 MHz; ¹³C: 100 MHz) were recorded on a Bruker 400 MHz spectrometer using DMSO-d₆ as the solvent.
4.2. Synthetic procedure compounds 2a-e
A mixture of commercially available benzaldehydes 1a-e (1 equiv) and 50% hydroxylamine solution (2 equiv) in ethanol was refluxed at 60 °C for 2 h. After cooling to room temperature, the precipitated solid was filtered off and washed with ethanol to offer derivatives 2a-e as white solid.
4.3. Synthetic procedure compounds 3a-e
A mixture of derivatives 2a-e (1 equiv) and N-Chlorosuccinimide (5 equiv) in DMF was refluxed at 40 °C for 2 h. Upon completion of the reaction indicated by TLC, the mixture was quenched by water, and subsequently was extracted with ethyl acetate for three times. The combined organic layers were evacuated to provide the residue, which were recrystallized with ethyl acetate, producing title compounds 3a-e.
4.4. Synthetic procedure compounds 4a-e
A mixture of derivatives 4a-e (1 equiv), triethylamine (2 equiv) and methyl 3-cyclopropyl-3-oxopropionate (1 equiv) in ethanol was refluxed at room temperature for 5–6 h. Upon completion of the reaction indicated by TLC, the mixture was quenched by water, and subsequently was extracted with ethyl acetate for three times. The combined organic layers were evacuated to provide the residue, which were recrystallized with ethyl acetate, producing title compounds 4a-e.
4.5. Synthetic procedure compounds 5a-e
A mixture of derivatives 4a-e (1 equiv) and sodium hydroxide (3 equiv) in water was refluxed at 80 °C for 1 h. Upon completion of the reaction indicated by TLC, the resulting mixture was acidified with 0.5 N aq. HCl to pH 7–8. The precipitate was filtered, washed with water, recrystallized with ethanol and dried under reduced pressure to provide derivatives 5a-e.
4.6. Synthetic procedure compounds 6a-p
A solution of 5a-e (1 equiv), different substituted amines (1 equiv), N,N-diisopropylethylamine (1 equiv), and EDCI (1 equiv) in dichloromethane was reacted at room temperature for 2 h. Upon completion of the reaction indicated by TLC, the organic layers were evacuated to offer the residue, which was subsequently purified by chromatography (silica gel, 5% MeOH/DCM for 6a-f, 6% MeOH/DCM for 6g, 8% MeOH/DCM for 6h-l, 7% MeOH/DCM for 6m-p,) to offer compounds 6a-p.
4.7. Synthetic procedure compounds 7–22
A solution of compounds 6a-p (1 equiv), sodium hydroxide (1 equiv), and 50% hydroxylamine solution (2 equiv) in methanol were reacted at room temperature for 1 h. Upon completion of the reaction indicated by TLC, the resulting mixture was evacuated to offer the residue, which was acidified with 0.5 N aq. formic acid to pH 6–7. The precipitate was subjected to filtration, followed by washing with water. It was then recrystallized using methanol and subsequently dried under reduced pressure, resulting in the formation of title compounds 7–22.
3-(4-chlorophenyl)-5-cyclopropyl-N-(2-(hydroxyamino)-2-oxoethyl)isoxazole-4-carboxamide (7). Yield 68%. White solid. Mp: 156.6–157.5 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.80 (t, J = 6.0 Hz, 1H), 8.49 (s, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.1 Hz, 2H), 3.78 (d, J = 5.8 Hz, 2H), 1.28–1.01 (m, 5H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.85, 166.21 (d, J = 58.3 Hz), 162.22, 159.73, 135.30, 129.77 (d, J = 92.0 Hz), 127.43, 112.26, 9.21, 8.36. MS (ESI), calcd. C_15_H_14_ClN_3_O_4_, [M + H] + m/z: 338.41. found: 338.42.
3-(4-chlorophenyl)-5-cyclopropyl-N-(3-(hydroxyamino)-3-oxopropyl)isoxazole-4-carboxamide (8). Yield 65%. White solid. Mp: 177.4–178.2 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 8.88 (s, 1H), 8.58 (t, J = 5.7 Hz, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.3 Hz, 2H), 3.42 (q, J = 6.6 Hz, 2H), 2.33 (td, J = 8.6, 4.4 Hz, 1H), 2.25 (t, J = 7.0 Hz, 2H), 1.15 (dq, J = 9.8, 6.0, 4.5 Hz, 2H), 1.09 (dd, J = 5.2, 2.2 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.59, 167.46, 161.78, 159.66, 135.31, 129.72 (d, J = 60.3 Hz), 127.49, 112.61, 36.33, 32.44, 8.99, 8.28. MS (ESI), calcd. C_16_H_16_ClN_3_O_4_, [M + H] + m/z: 350.21. found: 350.22.
3-(4-chlorophenyl)-5-cyclopropyl-N-(4-(hydroxyamino)-4-oxobutyl) isoxazole-4-carboxamide (9). Yield 66%. White solid. Mp: 136.8–137.6 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.51 (t, J = 5.5 Hz, 1H), 7.68 (d, J = 8.3 Hz, 2H), 7.59 (d, J = 8.2 Hz, 2H), 3.21 (q, J = 6.5 Hz, 2H), 2.45–2.27 (m, 1H), 2.12 (s, 1H), 2.01 (t, J = 7.5 Hz, 1H), 1.71 (t, J = 7.3 Hz, 2H), 1.17 (dd, J = 8.0, 4.5 Hz, 2H), 1.09 (dt, J = 8.4, 3.7 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.35, 169.17, 161.74, 159.68, 135.36, 129.70 (d, J = 57.0 Hz), 127.55, 112.77, 30.41, 25.51, 8.59 (d, J = 62.2 Hz). MS (ESI), calcd. C_17_H_18_ClN_3_O_4_, [M + Na] + m/z: 386.19. found: 386.20.
3-(4-chlorophenyl)-5-cyclopropyl-N-(5-(hydroxyamino)-5-oxopentyl) isoxazole-4-carboxamide (10). Yield 67%. White solid. Mp: 124.3–125.7 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.51 (t, J = 5.8 Hz, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.1 Hz, 2H), 3.34 (s, 2H), 3.21 (q, J = 6.3 Hz, 2H), 2.29 (tt, J = 8.4, 4.9 Hz, 1H), 1.98 (t, J = 7.0 Hz, 1H), 1.49 (dq, J = 22.0, 7.6 Hz, 4H), 1.17 (dq, J = 7.6, 4.2 Hz, 2H), 1.09 (dq, J = 7.8, 4.9 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.22, 169.38, 161.69, 159.65, 135.36, 129.68 (d, J = 49.6 Hz), 127.56, 112.87, 32.34, 28.87, 23.12, 8.84, 8.27. MS (ESI), calcd. C_18_H_20_ClN_3_O_4_, [M + Na] + m/z: 400.17. found: 400.16.
3-(4-chlorophenyl)-5-cyclopropyl-N-(4-(hydroxycarbamoyl) benzyl) isoxazole-4-carboxamide (11). Yield 63%. White solid. Mp: 210.5–211.7 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 9.03 (t, J = 6.0 Hz, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.58–7.45 (m, 2H), 7.35 (d, J = 8.0 Hz, 2H), 4.46 (d, J = 5.9 Hz, 2H), 2.32 (s, 1H), 1.17 (dq, J = 8.3, 3.3, 2.8 Hz, 2H), 1.10 (dt, J = 5.6, 3.0 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.57, 164.33, 161.93, 159.79, 142.39, 135.37, 132.10, 130.06, 129.34, 127.88, 127.44 (d, J = 3.6 Hz), 112.56, 42.99, 8.95, 8.31. MS (ESI), calcd. C_21_H_18_ClN_3_O_4_, [M + H] + m/z: 412.21. found: 412.20.
5-cyclopropyl-N-(4-(hydroxyamino)-4-oxobutyl)-3-(p-tolyl)isoxazole-4-carboxamide (12). Yield 66%. White solid. Mp: 142.5–146.3 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.51 (d, J = 5.8 Hz, 1H), 8.47 (d, J = 15.6 Hz, 1H), 7.55 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 7.8 Hz, 2H), 3.19 (q, J = 6.7 Hz, 2H), 2.37 (s, 3H), 2.32–2.20 (m, 1H), 2.00 (t, J = 7.5 Hz, 1H), 1.79–1.60 (m, 2H), 1.15 (dt, J = 8.4, 3.2 Hz, 2H), 1.07 (dq, J = 7.7, 4.9, 4.2 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 172.74, 169.10, 166.35, 162.05, 160.42, 140.19, 129.81, 128.01, 125.85, 112.80, 30.41, 25.54, 21.43, 8.70, 8.22. MS (ESI), calcd. C_18_H 21_N_3_O_4, [M + H] + m/z: 344.32. found: 344.31.
5-cyclopropyl-3-(4-fluorophenyl)-N-(4-(hydroxyamino)-4-oxobutyl)isoxazole-4-carboxamide (13). Yield 62%. White solid. Mp: 115.3–116.2 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.51 (s, 1H), 8.40 (s, 1H), 7.66–7.56 (m, 2H), 7.06 (d, J = 120.8 Hz, 2H), 3.83 (s, 3H), 2.33–2.19 (m, 1H), 2.01 (t, J = 7.5 Hz, 1H), 1.78–1.66 (m, 2H), 1.14 (dt, J = 8.3, 3.2 Hz, 2H), 1.07 (dt, J = 5.6, 3.1 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 172.62, 169.15, 165.86, 162.15, 161.06, 160.06, 157.49, 129.56, 120.92, 114.72, 112.67, 55.79, 30.42, 25.55, 8.67, 8.21. MS (ESI), calcd. C_17_H 18_FN_3_O_4, [M + Na] + m/z: 370.24. found: 370.25.
5-cyclopropyl-N-(2-(hydroxyamino)-2-oxoethyl)-3-(4-methoxyphenyl)isoxazole-4-carboxamide (14). Yield 65%. White solid. Mp: 149.7–150.2 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.88 (s, 1H), 8.67 (t, J = 6.0 Hz, 1H), 7.63 (d, J = 7.9 Hz, 2H), 7.28 (d, J = 7.9 Hz, 3H), 3.76 (d, J = 5.9 Hz, 2H), 2.52 (p, J = 1.9 Hz, 2H), 2.47 (d, J = 10.0 Hz, 1H), 1.13 (ddt, J = 10.9, 7.9, 2.7 Hz, 5H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.34, 166.04, 162.50, 160.48, 140.11, 129.77, 128.25, 125.72, 112.22, 21.44, 9.06, 8.30. MS (ESI), calcd. C_16_H 17_N_3_O_5, [M + Na] + m/z: 354.23. found: 354.23/338.23.
5-cyclopropyl-N-(3-(hydroxyamino)-3-oxopropyl)-3-(4-methoxyphenyl)isoxazole-4-carboxamide (15). Yield 63%. White solid. Mp: 174.5–175.2 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.53 (t, J = 5.7 Hz, 1H), 8.42 (s, 1H), 7.80 (s, 1H), 7.54 (d, J = 7.9 Hz, 2H), 7.30 (d, J = 7.9 Hz, 2H), 3.41 (q, J = 6.7 Hz, 2H), 2.52 (p, J = 1.8 Hz, 2H), 2.36 (s, 1H), 2.23 (d, J = 7.2 Hz, 2H), 1.24–1.10 (m, 2H), 1.06 (dq, J = 7.7, 5.0, 4.3 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 172.98, 167.40, 166.07, 162.07, 160.44, 157.46, 140.12, 129.82, 128.08, 125.80, 112.65, 36.34, 32.49, 21.44, 8.82, 8.22. MS (ESI), calcd. C_17_H 19_N_3_O_5, [M + Na] + m/z: 368.23. found: 352.23/368.23.
5-cyclopropyl-N-(4-(hydroxyamino)-4-oxobutyl)-3-(4- methoxyphenyl) isoxazole-4-carboxamide (16). Yield 65%. White solid. Mp: 158.8–159.7 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.50 (t, J = 5.7 Hz, 1H), 8.40 (d, J = 1.8 Hz, 1H), 7.67–7.49 (m, 2H), 7.17–6.82 (m, 2H), 3.82 (d, J = 1.4 Hz, 3H), 3.20 (q, J = 6.8 Hz, 2H), 2.36–2.17 (m, 1H), 2.01 (t, J = 7.5 Hz, 2H), 1.71 (q, J = 7.4 Hz, 2H), 1.24–1.14 (m, 2H), 1.06 (dt, J = 5.3, 3.3 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 172.62, 169.15, 165.87, 162.15, 160.56 (d, J = 100.8 Hz), 157.49, 129.55, 120.92, 113.69 (d, J = 206.5 Hz), 55.78, 30.42, 25.55, 8.66, 8.21. MS (ESI), calcd. C_18_H_21_N_3_O_5_, [M + Na] + m/z: 382.14. found: 382.15.
5-cyclopropyl-N-(5-(hydroxyamino)-5-oxopentyl)-3-(4-methoxyphenyl)isoxazole-4-carboxamide (17). Yield 68%. White solid. Mp: 142.5–143.7 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 8.58–8.34 (m, 1H), 7.60 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 3.82 (s, 3H), 3.20 (q, J = 6.3 Hz, 2H), 2.25 (dt, J = 8.4, 3.6 Hz, 1H), 1.98 (t, J = 7.0 Hz, 2H), 1.49 (dq, J = 21.6, 7.5 Hz, 4H), 1.14 (dd, J = 7.9, 3.3 Hz, 2H), 1.05 (dq, J = 7.7, 4.8, 4.3 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 172.52, 169.33, 166.62, 162.10, 161.06, 160.02, 129.51, 120.92, 114.73, 112.75, 55.78, 32.38, 28.89, 23.15, 8.63, 8.20. MS (ESI), calcd. C_19_H 23_N_3_O_5, [M + Na] + m/z: 396.21. found: 396.20.
5-cyclopropyl-N-(4-(hydroxycarbamoyl)benzyl)-3-(4-methoxyphenyl)isoxazole-4-carboxamide (18). Yield 63%. White solid. Mp: 205.5–206.3 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.53 (s, 1H), 7.71 (d, J = 7.8 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 7.9 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 4.42 (d, J = 5.9 Hz, 2H), 3.81 (s, 3H), 2.26 (s, 1H), 1.13 (dt, J = 8.5, 3.1 Hz, 2H), 1.06 (q, J = 3.5, 2.6 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 166.67, 129.60, 129.55, 129.20, 127.73, 127.04, 114.63, 114.12, 55.78, 8.67 (d, J = 2.8 Hz), 8.58, 8.22, 8.20. MS (ESI), calcd. C_22_H 21_N_3_O_5, [M + H] + m/z: 408.15. found: 408.16.
3-(4-cyanophenyl)-5-cyclopropyl-N-(2-(hydroxyamino)-2-oxoethyl)isoxazole-4-carboxamide (19). Yield 63%. White solid. Mp: 152.4–153.5 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.82 (t, J = 6.2 Hz, 1H), 8.48 (s, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.77 (dq, J = 17.6, 8.4, 7.9 Hz, 2H), 5.89 (s, 1H), 3.77 (d, J = 5.9 Hz, 2H), 2.52–2.42 (m, 1H), 1.26–1.15 (m, 2H), 1.14–1.01 (m, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.53, 166.34, 165.94, 162.39, 150.77, 135.24, 128.84, 128.32, 128.25, 128.11, 126.10, 9.18, 9.13, 8.34. MS (ESI), calcd. C_16_H 14_N_4_O_4, [M + K] + m/z: 367.06. found: 367.16.
3-(4-cyanophenyl)-5-cyclopropyl-N-(3-(hydroxyamino)-3-oxopropyl)isoxazole-4-carboxamide (20). Yield 67%. White solid. Mp: 173.2–174.5 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.63 (q, J = 5.5, 4.9 Hz, 1H), 8.49 (s, 1H), 7.98 (s, 1H), 7.90–7.81 (m, 1H), 7.81–7.67 (m, 1H), 7.65 (d, J = 8.1 Hz, 1H), 5.90 (s, 1H), 3.43 (q, J = 6.8 Hz, 2H), 2.42–2.27 (m, 1H), 2.25 (d, J = 7.3 Hz, 2H), 1.17 (dt, J = 8.5, 3.8 Hz, 2H), 1.08 (s, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 167.42, 166.46, 161.94, 150.80, 135.29, 128.20, 128.08, 127.95, 126.20, 112.71, 36.35, 32.48, 8.99, 8.92, 8.27. MS (ESI), calcd. C_17_H 16_N_4_O_4, [M + Na] + m/z: 374.26. found: 374.25.
3-(4-cyanophenyl)-5-cyclopropyl-N-(4-(hydroxyamino)-4-oxobutyl)isoxazole-4-carboxamide (21). Yield 63%. White solid. Mp: 182.4–183.3 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 8.57 (t, J = 5.6 Hz, 1H), 8.51 (d, J = 1.3 Hz, 1H), 7.97 (s, 1H), 7.83 (d, J = 19.4 Hz, 1H), 7.70 (dd, J = 17.6, 9.3 Hz, 1H), 7.64 (s, 1H), 5.90 (s, 1H), 3.21 (q, J = 6.7 Hz, 2H), 2.30 (td, J = 8.5, 4.3 Hz, 1H), 2.08–1.94 (m, 2H), 1.70 (q, J = 7.4 Hz, 2H), 1.17 (dq, J = 7.2, 3.7 Hz, 2H), 1.09 (dt, J = 6.6, 3.0 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d6) δ 169.11, 166.76, 161.78, 160.16, 160.07, 150.74, 128.36, 128.11, 128.02, 127.88, 127.69, 112.93, 30.42, 25.55, 8.87, 8.80, 8.26. MS (ESI), calcd. C_18_H 18_N_4_O_4, [M + K] + m/z: 395.09. found: 395.23.
3-(4-cyanophenyl)-5-cyclopropyl-N-(5-(hydroxyamino)-5-oxopentyl)isoxazole-4-carboxamide (22). Yield 67%. White solid. Mp: 208.0–209.5 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.69 (s, 1H), 8.50 (d, J = 5.9 Hz, 1H), 8.20–7.93 (m, 2H), 7.75 (dd, J = 28.9, 8.0 Hz, 2H), 3.21 (q, J = 6.4 Hz, 2H), 2.29 (dt, J = 8.7, 3.5 Hz, 1H), 1.98 (t, J = 7.0 Hz, 2H), 1.58–1.40 (m, 4H), 1.23–1.03 (m, 4H). ^13^C NMR (100 MHz, DMSO-d6) δ 173.17, 168.61 (d, J = 163.3 Hz), 161.73, 160.02, 136.08, 131.23, 128.36, 127.99, 127.85, 126.18, 113.02, 32.34, 28.85, 23.12, 8.83, 8.26. MS (ESI), calcd. C_19_H 20_N_4_O_4, [M + K] + m/z: 409.35. found: 409.34.
4.8. MTT assay
Cells were seeded in 96-well plates at a density of 2−3 × 10³ cells per well. On the following day, 200 μL of fresh complete medium containing serial concentrations of tested compounds was added to the plates, which were then incubated for 24 h, 48 h, and 72 h individually. For PC3 and WPMY-1 cells, pre-treatment with 4 mM SB203580 was performed for 4 h, followed by treatment with the IC_50_ concentration of the compounds for 72 h. Subsequently, 20 μL of MTT solution (5 mg/mL, dissolved in PBS) was added to each well. After an additional 4 h incubation at 37°C in the dark, absorbance was measured at 490 nm using a microplate reader. The absorbance values were analyzed, and IC_50_ values were calculated using SPSS software.
4.9. HDAC1 inhibition assay
A 100 μL mixture of HDAC1 solution and compounds at different concentrations was incubated in assay buffer (2 mM MgCl₂; 2 mM KCl; 100 mM NaCl; 20 mM HEPES, pH 7.5) at 37°C for a duration of 2 minutes. Next, the substrate Boc-LGK-Acetyl-AMC was added to achieve a final concentration of 1 mM, and the reactions were allowed to proceed at 37°C for an additional 20 minutes. To terminate the reactions, trypsin was introduced, followed by a subsequent incubation period of 15 minutes at 37°C. Fluorescence intensity was determined with a microplate reader with excitation set to 355 nm and emission detected at 460 nm. Inhibitory ratios were calculated utilizing Prism version 6.0 software [23].
4.10. Molecular docking
Molecular docking studies were conducted utilizing the Molecular Operating Environment (MOE) software. The crystal structure of HDAC1 (PDB ID: 5ICN) was employed as the docking receptor [24]. Receptor preparation involved the addition of missing atoms, removal of water molecules, and energy minimization with the QuickPrep module. Three-dimensional structures for derivatives 7 and 17 were generated via conformational searching and subsequent energy minimization. The protonation states of both ligand and receptor were adjusted to a pH of 7.0. During the docking process, HDAC1 protein was maintained in a rigid conformation while allowing flexibility for small molecules. The top 20 scoring poses were selected for analysis. The binding mode visualizations were produced through PyMOL [25].
Supporting information
S1 FileCompounds 7–22 characterization.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Carreiras M do C, Marco-Contelles J. Hydrazides as Inhibitors of Histone Deacetylases. J Med Chem. 2024;67(16):13512–33. doi: 10.1021/acs.jmedchem.4c 00541 39092855 · doi ↗ · pubmed ↗
- 2Geng H, Zheng F, Sun W, Huang S, Wang Z, Yang K, et al. Effect and mechanism of novel HDAC inhibitor ZDLT-1 in colorectal cancer by regulating apoptosis and inflammation. Int Immunopharmacol. 2024;143(Pt 1):113333. doi: 10.1016/j.intimp.2024.113333 39383785 · doi ↗ · pubmed ↗
- 3Xu T, Fang Y, Gu Y, Xu D, Hu T, Yu T, et al. HDAC inhibitor SAHA enhances antitumor immunity via the HDAC 1/JAK 1/FGL 1 axis in lung adenocarcinoma. J Immunother Cancer. 2024;12(10):e 010077. doi: 10.1136/jitc-2024-010077 39384195 PMC 11474878 · doi ↗ · pubmed ↗
- 4Kraft FB, Biermann L, Schäker-Hübner L, Hanl M, Hamacher A, Kassack MU, et al. Hydrazide-Based Class I Selective HDAC Inhibitors Completely Reverse Chemoresistance Synergistically in Platinum-Resistant Solid Cancer Cells. J Med Chem. 2024;67(19):17796–819. doi: 10.1021/acs.jmedchem.4c 01817 39356226 · doi ↗ · pubmed ↗
- 5Cao D-F, Zhou X-Y, Guo Q, Xiang M-Y, Bao M-H, He B-S, et al. Unveiling the role of histone deacetylases in neurological diseases: focus on epilepsy. Biomark Res. 2024;12(1):142. doi: 10.1186/s 40364-024-00687-6 39563472 PMC 11575089 · doi ↗ · pubmed ↗
- 6Xie J, Liu R, Cai Y, Liu D. HDAC 1: a promising target for cancer treatment: insights from a thorough analysis of tumor functions. Transl Cancer Res. 2024;13(10):5300–15. doi: 10.21037/tcr-24-23 39525004 PMC 11543092 · doi ↗ · pubmed ↗
- 7Jenke R, Oliinyk D, Zenz T, Körfer J, Schäker-Hübner L, Hansen FK, et al. HDAC inhibitors activate lipid peroxidation and ferroptosis in gastric cancer. Biochem Pharmacol. 2024;225:116257. doi: 10.1016/j.bcp.2024.116257 38705532 · doi ↗ · pubmed ↗
- 8Wang L, Bai Y, Cao Z, Guo Z, Lian Y, Liu P, et al. Histone deacetylases and inhibitors in diabetes mellitus and its complications. Biomed Pharmacother. 2024;177:117010. doi: 10.1016/j.biopha.2024.117010 38941890 · doi ↗ · pubmed ↗