Severe Neurological Presentation in Siblings With COQ5 ‐Related Primary Coenzyme Q10 Deficiency: Expanding Clinical and Molecular Spectrum
Parith Wongkittichote, Rachel M. Guerra, Daniel J. Wegner, Samantha Toy, Jacqueline A. Hauer, David J. Pagliarini, Jorge L. Granadillo

TL;DR
This paper reports two siblings with severe neurological issues caused by a genetic mutation in COQ5, a gene involved in CoQ10 production, and shows that CoQ10 supplementation may help.
Contribution
The study expands the clinical and molecular spectrum of COQ5-related CoQ10 deficiency and establishes a yeast model to evaluate COQ5 variants.
Findings
Compound heterozygous COQ5 variants were identified in two siblings with severe neurological symptoms.
A yeast model showed that a missense COQ5 variant failed to rescue CoQ biosynthesis, leading to intermediate accumulation.
CoQ10 supplementation led to subjective clinical improvement in the proband.
Abstract
Coenzyme Q10 (CoQ10) is a coenzyme and antioxidant involved in multiple bioenergetic and biosynthetic processes, particularly within mitochondria. The biosynthesis of CoQ10 is a tightly regulated process that involves multiple enzymes, including the methyltransferase COQ5. Genetic defects in COQ5 have recently been associated with autosomal recessive COQ5‐related primary CoQ10 deficiency. The clinical manifestations of seven individuals previously reported were primarily neurological and ophthalmological. Here, we report two siblings with profound developmental delay and brain imaging consistent with multistage strokes. Clinical exome sequencing revealed compound heterozygous variants in COQ5, including one frameshift deletion and one missense variant. Our functional complementation studies demonstrate that a Saccharomyces cerevisiae COQ5 ortholog harboring the corresponding missense…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
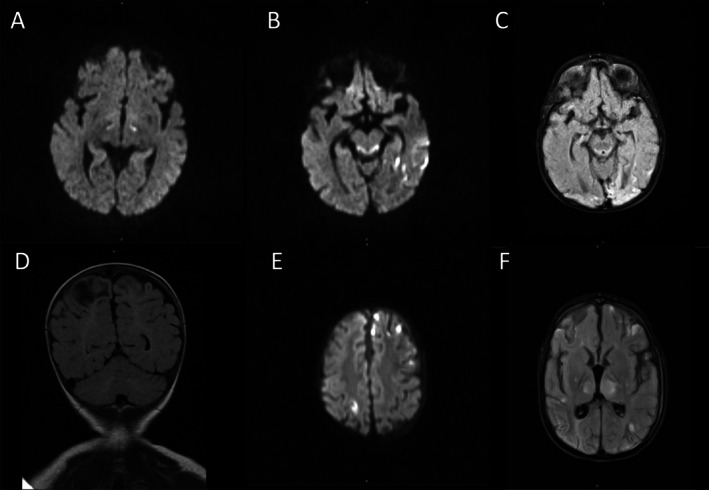 FIGURE 1
FIGURE 1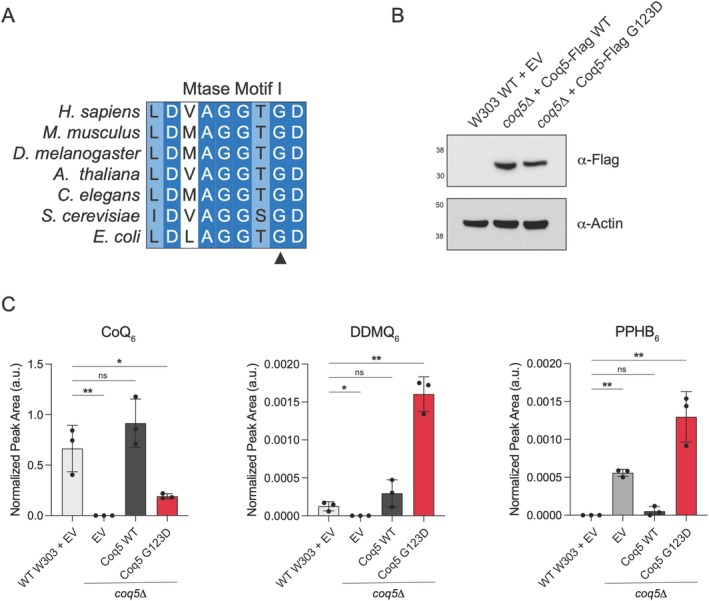 FIGURE 2
FIGURE 2| c.353G>A (p.Gly118Asp) | |
|---|---|
| Splice AI | Splice‐Altering/moderate (0.25) |
| CADD | 33 |
| Revel | Deleterious (0.97) |
| Varity | Deleterious (0.99) |
| SIFT | Deleterious (0) |
| MT | Deleterious (1) |
| ACMG criteria | PS3, PM2, PP3, PP5 |
| Interpretation | Pathogenic |
| Patient | Najmabadi et al. [ | Malicdan et al. [ | Dawidziuk et al. [ | Jurkute [ | Present study | ||||
|---|---|---|---|---|---|---|---|---|---|
| M144 | III.4 | III.3 | III.6 | Family 11 | Family 12 | Patient 1 | Patient 2 | ||
| Variant | c.352G>A (p.Gly118Ser) | c.575‐1761_*1489dup (p.?) | c.575‐1761_*1489dup (p.?) | c.575‐1761_*1489dup (p.?) |
c.352G>A (p.Gly118Ser) c.681 + 1G>A (p.?) |
c.933delC (p.Tyr311*) c.682–7>G |
c.367C>T (p.Arg123Trp) c.682–7>G |
c.177_178del (p.Ser60Glyfs13) c.353G>A (p.Gly118Asp) |
c.177_178del (p.Ser60Glyfs13) c.353G>A (p.Gly118Asp) |
| Zygosity | Homozygous | Homozygous | Homozygous | Homozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous |
| Ethnicity | N/A | Iraqi‐Jewish | Iraqi‐Jewish | Iraqi‐Jewish | Polish | N/A | N/A | White | White |
| Gender | N/A | Female | Female | Female | Female | Male | Female | Female | Male |
| Consanguinity | Yes | No | No | No | No | N/A | N/A | No | No |
| Age at onset | N/A | N/A | N/A | N/A | 5 m | N/A | N/A | 6 m | 3 m |
| Age at examination | N/A | N/A | N/A | N/A | 10 y | 56 y | 38 y | 27 months | 7 years |
| Neurologic | |||||||||
| Developmental delay (mild/moderate/severe) | Severe | Moderate | None | Mild | Severe | — | — | Severe | Severe |
| Intellectual disability | N/A | N/A | Borderline | Mild | Yes | — | — | N/A | N/A |
| Microcephaly | N/A | N/A | N/A | N/A | Yes | — | — | Yes | Yes |
| Spasticity | N/A | Yes (LL) | N/A | N/A | Yes (LL) | — | — | Yes | Yes |
| Stroke‐like | N/A | — | — | — | — | — | — | Yes | Yes |
| Ataxia | N/A | Yes | Yes | Yes | N/A | — | — | N/A | N/A |
| Seizure | N/A | Yes | Yes | Yes | No | — | — | Yes | Yes |
| Dysarthria | N/A | Yes | Yes | Yes | Pronounce a few simple syllables | — | — | Nonverbal | Nonverbal |
| Ophthalmologic (optic nerve atrophy, retinopathy, cataract, strabismus, ptosis, ie) | N/A | Horizontal nystagmus, slow saccades with saccadic pursuit and apraxic gaze | Oculomotor apraxia, nystagmus | Horizontal nystagmus, hypometric saccades. | Normal | RP | RP, nystagmus | RP, optic atrophy | Esotropia |
| Other clinical manifestations | N/A | Short stature, impulsivity, attention deficit, oppositional characteristics | N/A | N/A | Short stature, impulsivity, attention deficit | Hypertension | Weakness, Under developed fertile function | Feeding difficulties | Feeding difficulties |
| Brain MRI | N/A | Mild non‐progressive cerebellar atrophy | Mild cerebellar atrophy | N/A | Progressive cerebellar atrophy | N/A | N/A | Abnormal signal intensity in bilateral medial thalami and dorsal midbrain, recurrent strokes | Encephalomalacia, hyperintensities in the bilateral thalami and brainstem, multiple strokes of varying age |
| Respiratory chain enzymes | N/A | Decreased Complex II + III activity | N/A | N/A | Normal | N/A | N/A | N/A | N/A |
| CoQ10 level (normal) | Leukocyte: 65 pmol/ug protein (119.86 ± 24.23); Muscle 21.53 μg/g tissue (26.63–48.11) | Leukocyte: 78 pmol/μg protein (119.86 ± 24.23) | Leukocyte: 72 pmol/μg protein (119.86 ± 24.23) | Plasma: 0.6 mg/L (> 0.67 mg/L) | Plasma: 582.28 nmol/L (227–1432) | None | Leukocyte: 38 pmol/mg protein (66–183 pmol) | Leukocyte: 39 pmol/mg protein (66–183 pmol) | |
| Treatment | N/A | CoQ10 15 mg/kg/day | CoQ10 15 mg/kg/day | CoQ10 15 mg/kg/day | QuinoMit Q10 Fluid at 26 mg every second day | None | None | CoQ10 15 mg/kg/day | None |
- —National Institute of General Medical Sciences10.13039/100000057
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCoenzyme Q10 studies and effects · Mitochondrial Function and Pathology · Metalloenzymes and iron-sulfur proteins
Summary
- This study expands the clinical spectrum of COQ5‐related primary CoQ_10_ deficiency to include severe neurological abnormalities with multi‐stage strokes.
- It also validates a yeast‐based functional assay using targeted lipidomics, providing a valuable tool for characterizing human COQ5 variants.
Introduction
1
Coenzyme Q_10_ (CoQ_10_), composed of a benzoquinone head group and a long polyisoprenyl tail, is a coenzyme and antioxidant used in multiple cellular processes, including the mitochondrial electron transport chain, pyrimidine biosynthesis, ferroptosis defense, and the maintenance of cellular redox balance [1, 2, 3]. The biosynthesis of CoQ_10_ is a tightly regulated multistep process involving multiple enzymes [2]. Defects in CoQ_10_ biosynthesis have been associated with a rare group of heterogeneous disorders, namely primary CoQ_10_ deficiency [4, 5]. They share overlapping features including developmental delay, encephalopathy, epilepsy, ataxia, and myopathy. Additional presentations include steroid‐resistant nephrotic syndrome, hypertrophic cardiomyopathy, optic atrophy, and retinopathy [6, 7]. The patients may exhibit abnormal biochemical analysis including reduced CoQ_10_ levels in muscle, fibroblast, or leukocytes, or abnormal mitochondrial transport enzymes activity [6].
To date, at least 13 enzymes are known to be associated with CoQ_10_ metabolism [2, 4]. Briefly, 4‐hydroxybenzoic acid (4‐HB), generated from tyrosine, is linked to polyisoprenyl pyrophosphate to form 3‐hexaprenyl‐4‐hydroxy benzoic acid (PPHB), which is further modified by multiple enzymes, including an S‐adenosylmethionine‐dependent methyltransferase encoded by COQ5 (MIM# 616359), located on chromosome 12q24.31. In Baker's yeast ( Saccharomyces cerevisiae ), the knock‐out of the human orthologue COQ5 (coq5Δ) displayed an oxidative growth defect and accumulation of its substrate, demethyl‐demethoxy‐Q_6_ (DDMQ_6_) [8].
COQ5‐related primary CoQ_10_ deficiency (COQ5‐PCD) is an extremely rare autosomal recessive disorder that was initially described in three siblings born to non‐consanguineous parents of Iraqi‐Jewish descent who presented with cerebellar ataxia, epilepsy, developmental, and cognitive delay [9]. Biochemical studies showed reduced leukocyte and muscle CoQ_10_ levels. Genetic testing revealed a homozygous exon duplication causing an abnormally long 3′ untranslated region (3′UTR). Four additional cases have been reported with a combination of missense, splicing, and truncating variants [10, 11, 12]. These findings indicate that loss of function is a presumptive molecular mechanism. However, these studies did not assess the effect of these variants on CoQ_10_ metabolism. Moreover, the lack of a functional assay made assessment of missense variants challenging.
Here we identified two novel variants in COQ5 in two siblings with profound developmental delay, epilepsy, hypotonia, and stroke‐like episodes via clinical exome sequencing. We also employ complementation studies in S. cerevisiae lacking a Coq5 ortholog (coq5∆) to analyze the biochemical effects of this missense variant. We reveal that the yeast Coq5 ortholog harboring the corresponding mutation fails to rescue CoQ_6_ levels (the S. cerevisiae equivalent of human CoQ_10_) and results in the accumulation of CoQ_6_ intermediates, thereby supporting the likely pathogenicity of this variant.
Materials and Methods
2
Case Description
2.1
Patient 1
2.1.1
The proband was a 4‐year‐old white female who was born to a 26‐year‐old mother. Pregnancy was uncomplicated. She was born at 39 weeks 1 day of estimated gestational age (EGA) via planned cesarean section. Delivery and neonatal courses were uneventful. Her birth weight was at the 25^th^ percentile, birth length was at the 59^th^ percentile, and occipitofrontal circumference (OFC) was at the 4^th^ percentile. She was noted to have developmental delay at 6 months of age. Family history was notable for three healthy siblings and a brother who also had developmental delay (Patient 2). Brain magnetic resonance imaging (MRI) at 21 months of age showed reduced diffusivity within the bilateral ventral thalami and cerebral peduncles with diffuse nonspecific white matter volume loss (Figure 1A). Follow‐up brain MRI at 25 months of age showed worsening signal abnormality within the dorsal midbrain and medial thalami and new abnormality involving the left PCA territory suggestive of subacute infarct (Figure 1B). Head and neck magnetic resonance angiography (MRA) did not detect significant stenosis or occlusion. Brain MRI at 33 months showed gliosis of previously demonstrated areas of ischemic injury in thalami, brainstem, and left dorsal cerebral cortices, and new areas of late subacute to chronic ischemic injury in right posterior temporal and occipital cortices (Figure 1C).
Brain imaging of the probands. (A–C) Brain MRI of Patient 1: (A) Brain MRI at the age of 21 months of age demonstrated reduced diffusivity in bilateral thalami on diffusion weighted imaging (B) At the age of 25 months, brain MRI showed reduced diffusivity in bilateral dorsal midbrain and left dorsal cerebral cortices on diffusion weighted imaging. (C) At 33 months of age, brain MRI showed gliosis in brainstem and left dorsal cerebral cortices as well as evolving ischemic injury in right dorsal cerebral cortices on T2/FLAIR imaging. (D–F) Brain MRI of Patient 2: (D) Brain MRI at 3 months of age demonstrated encephalomalacia in right greater than left parietal cortices on T2/FLAIR imaging. (E–F) Brain MRI at 3 years of age showed multifocal distribution of decreased diffusivity on diffusion weighted imaging (E) and hyperintensity on T2/FLAIR imaging (F).
She developed new onset seizure at 26 months of age. Electroencephalography (EEG) revealed abnormal spike and wave complexes predominantly in the left temporooccipital head region and synchronously in the right temporoparietooccipital head region. Later EEG at 60 months of age showed multifocal epileptiform discharges and generalized seizures. Repeated brain MRI showed stable abnormal signal intensity in bilateral medial thalami and dorsal midbrain and mild decrease in the edema associated with the suspected subacute infarct. She also had feeding difficulties and underwent gastrostomy tube placement at the age of 20 months due to failure to gain appropriate weight.
Upon evaluation at 27 months, she was able to roll over but could not sit or stand independently. She was able to reach out and transfer objects but could not use a pincer grasp. She could smile and coo but was nonverbal. Her weight and OFC were both below the 1^st^ percentile, with Z‐scores of −6.07 and −2.89, respectively. Her height was at the 1st percentile. Physical examination was notable for microcephaly and bilateral epicanthal folds but otherwise nondysmorphic. Neurologic examination was notable for sluggish pupillary reaction, axial hypotonia with spasticity of lower extremities, and generalized hyperreflexia. Ophthalmologic examination at the age of 4 years revealed ptosis, pale optic discs concerning for optic atrophy, and abnormal electroretinography consistent with retinopathy.
She underwent extensive metabolic evaluation including unremarkable total and free carnitine, acylcarnitine profile, serum amino acids, homocysteine, urine organic acids, creatine kinase, biotinidase, peroxisomal profile, and ammonia. Lactate was elevated with a maximum of 4.4 mmol/L (reference range 0.6–2.0 mmol/L) with a normal lactate: pyruvate ratio. Mitochondrial electron transport enzymes analysis in fibroblasts was normal. CoQ_10_ level in leukocytes was reduced to 38 pmol/mg protein (normal range 66–183 pmol/mg). Chromosomal microarray was normal.
At 37 months of age, supplementation with 15 mg/kg/day of CoQ10 was initiated. Within 2 months, her parents noted subjective clinical improvement in social interaction. Brain MRI at 40 and 45 months showed no new infarctions. From 42 months of age, CoQ10 supplementation was inconsistently given and eventually stopped. Brain MRI at 60 months showed new infarctions in the right posterior temporal occipital cortices.
Patient 2
2.1.2
Patient 2 is an elder brother of Patient 1. He was 7 years of age at the time of evaluation. He first presented with seizures at 3 months of age. Brain MRI at that time showed evolving encephalomalacia in bilateral parietal lobes (Figure 1D). EEG showed epileptiform discharges in the right central head region as well as clinicoelectrographic and subclinical seizures with onset in the same region. At 3 years of age, he presented with recurrent lactic acidosis and strokes in the setting of dehydration. Brain MRI at that time showed multiple strokes of varying ages, from acute/subacute to chronic (Figure 1E,F). It also showed hyperintensities in the bilateral thalami, as well as the brainstem, without corresponding diffusionopathy. He had a history of cerebral palsy, epilepsy, and profound intellectual disability. He was gastrostomy tube dependent, non‐verbal, and did not walk.
Unfortunately, he died at home at age 7 years due to unknown causes. Regarding laboratory evaluations, his lactate was normal. Leukocyte CoQ_10_ level was reduced to 39 pmol/mg protein (normal range 66–183 pmol/mg).
Clinical quad exome sequencing of Patient 1, Patient 2, and their parents revealed compound heterozygous variants in COQ5 (NM_032314.4) in both individuals: paternally inherited 2‐bp deletion, designated as c.177_178del (p.Ser60Glyfs13) and a maternally inherited missense variant, namely c.353G>A (p.Gly118Asp).
Next Generation Sequencing
2.2
Clinical exome sequencing was performed by GeneDx (Gaithersburg, MD). Bioinformatic analysis was performed based on the company's pipeline. In silico analysis of the effect of variants was performed by various bioinformatic tools including Splice AI [13], Combined Annotation Dependent Depletion (CADD) [14], Rare exome variant ensemble learner (Revel) [15], VARITY [16], Sorting Intolerant From Tolerant (SIFT) [17], MutationTaster2 (MT) [18] and Functional Analysis through Hidden Markov Models (FATHMM) [19]. Variant classification was performed according to the American College of Medical Genetics and Genomics (ACMG) recommendation [20].
Yeast Strains and Cultures
2.3
The S. cerevisiae haploid strain W303 (Matα; leu2‐3; trp1‐1; can1‐100; ura3‐1; ade2‐1; his3‐11) was used. The coq5Δ strain with deletion of yeast COQ5 was generated using PCR‐based homologous recombination, replacing the open reading frame of the targeting gene with the NatMX6 cassette [21]. The p416 plasmid with S. cerevisiae Coq5‐Flag expressed under a GPD promoter [22] was used as a template to generate the p.Gly123Asp point mutation via site‐directed mutagenesis. W303 WT and coq5Δ strains were transformed with p416 empty vector, p416‐GPD‐Coq5‐Flag WT, or p416‐GPD‐Coq5‐Flag G123D, using the LiAc/SS carrier DNA/PEG method [23] and plated on synthetic deficient (SD) Ura^−^ media with 2% glucose (US Biological). For lipid extraction and immunoblotting, 5 mL cultures of SD Ura, 2% glucose were inoculated with individual colonies and incubated (30°C, 230 r.p.m., 14–16 h). Cell density was measured as an optical density at 600 nm (OD_600_) and converted to cells mL^−1^ (1 OD = 1 × 10^7^ cells mL^−1^). 1 × 10^8^ cells were collected, pelleted, and snap‐frozen in LN_2_, and stored at −80°C.
Lipid Extraction and LC–MS Lipidomics
2.4
For lipid extractions, frozen cell pellets were thawed on ice, then 150 mM KCl (50 μL) was added to each sample, followed by ice‐cold methanol (600 μL; with 1 μM CoQ_8_ as internal standard). Glass beads (100 μL; 0.5 mm; BioSpec) were added, and the samples were vortexed (10 min, 4°C) to lyse the cells. Ice‐cold petroleum ether (400 μL) was added to extract the lipids, and the samples were vortexed again (3 min, 4°C). Samples were centrifuged (1000 × g, 3 min, RT) and the top petroleum ether layer was collected in a new tube. The petroleum ether extraction was repeated a second time, with the petroleum ether layer from the second extraction combined with that from the first. The extracted lipids were dried under argon before being resuspended in 2‐propanol (50 μL) and transferred to an amber glass vial (Sigma; QSertVial, 12 × 32 mm, 0.3 mL).
LC–MS analysis was performed using a Thermo Vanquish Horizon UHPLC system coupled to a Thermo Exploris 240 Orbitrap mass spectrometer. For LC separation, a Vanquish binary pump system (Thermo Fisher Scientific) was used with a Waters Acquity CSH C18 column (100 × 2.1 mm, 1.7 μm particle size) held at 35°C under a 300 μL/min flow rate. Mobile phase A consisted of 5 mM ammonium acetate in acetonitrile:H_2_O (70:30, v/v) with 125 μL/L acetic acid. Mobile phase B consisted of 5 mM ammonium acetate in isopropanol: acetonitrile (90:10, v/v) with the same additive. For each sample run, mobile phase B was initially held at 2% for 2 min and then increased to 30% over 3 min. Mobile phase B was further increased to 50% over 1 min and 85% over 14 min and then raised to 99% over 1 min and held for 4 min. The column was re‐equilibrated for 5 min at 2% B before the next injection. Five microliters of the sample were injected by a Vanquish Split Sampler HT autosampler (Thermo Fisher Scientific), while the autosampler temperature was kept at 4°C. The samples were ionized by a heated ESI source kept at a vaporizer temperature of 350°C. Sheath gas was set to 50 units, auxiliary gas to 8 units, sweep gas to 1 unit, and the spray voltage was set to 3500 V for positive mode and 2500 V for negative mode. The inlet ion transfer tube temperature was kept at 325°C with 70% RF lens. For targeted analysis, the MS was operated in parallel reaction monitoring mode with polarity switching acquiring scheduled, targeted scans to CoQ_6_ (m/z 591.4408), CoQ_8_ (m/z 727.566), and CoQ intermediates: DDMQ_6_ (m/z 547.4146) and PPHB_6_ (m/z 545.4). MS acquisition parameters include a resolution of 15,000, stepped HCD collision energy (25%, 30%, 40% for positive mode and 20%, 40%, 60% for negative mode), and 3 s dynamic exclusion. Automatic gain control targets were set to standard mode. The resulting CoQ intermediate data were processed using TraceFinder 5.1 (Thermo Fisher Scientific).
Western Blotting
2.5
Frozen yeast pellets were lysed in 150 μL of lysis buffer (1 M NaOH, 1 M BME) for 10 min with periodic vortexing. Protein was precipitated with 150 μL of 50% TCA and washed with 1 mL of acetone. The protein pellet was resuspended in 50 μL of 0.1 M NaOH and 25 μL of 3X LDS sample buffer. Proteins were separated (200 V, 35 min), transferred to a PVDF membrane (Sigma), and blocked with 5% non‐fat dry milk (1 h, RT.). Membranes were then probed with primary anti‐FLAG (Sigma, F1804, 1:1000) or anti‐actin (Sigma, 8224, 1:1000) antibodies (1 h, RT). Membranes were washed three times with TBST and then probed with HRP‐linked anti‐mouse IgG (Cell Signaling, #7076, 1:5000) secondary antibody (1 h, RT). Membranes were washed three times with TBST and developed using SignalFire ECL reagent (Cell Signaling). Developed membranes were imaged on a ChemiDoc system (Bio‐Rad).
Results
3
Molecular Analysis of
COQ5 Variants
3.1
Compound heterozygous variants in COQ5 (NM_032314.4) were detected in both patients: c.177_178del (p.Ser60Glyfs13) and c.353G>A (p.Gly118Asp). The 2‐bp deletion variant likely causes a frameshift, resulting in either nonsense mediated decay or protein truncation leading to the loss‐of‐function of the protein. In silico prediction of p.Gly118Asp supported a deleterious effect on enzyme function (Table 1). The base position c.353G is a splice acceptor site; however, in silico analysis for abnormal splicing was inconclusive [13]. We then performed an RNA study which revealed that the variant p.Gly118Asp does not affect splicing (Figure S1). None of the unaffected siblings are compound heterozygous for these two variants.
Functional Analysis Using Yeast Model System
3.2
Since the variant p.Gly118Asp does not cause splicing aberration, the effect of the variant on the protein had yet to be elucidated. We employed a Baker's yeast model to evaluate the functional impact of this variant using a homologous complementation approach. The residue p.Gly118 in human COQ5 corresponds to the conserved residue p.Gly123 in yeast COQ5 within the first methyltransferase motif (Figure 2A). Direct mutagenesis to the residue p.Gly123 was then performed to generate coq5 ^ G123D ^, which is equivalent to human p.Gly118Asp.
*Yeast COQ5
G123D does not support CoQ6 biosynthesis in coq5Δ. (A) Multiple sequence alignment of methyltransferase motif I of COQ5 homologs. Dark blue represents highly conserved residues, and light blue represents moderately conserved residues. The (▲) denotes human Gly118. (B) Western blotting of WT or coq5Δ yeast expressing EV or the indicated Coq5‐Flag construct under the control of the constitutive GPD promoter. (C) Relative total CoQ6, DDMQ6, and PPHB6 abundance of indicated Coq5‐Flag construct under the control of the constitutive GPD promoter. MS2‐based peak area is normalized to the CoQ8 internal standard. n = 3 biologically independent samples, two‐sided Student's t‐test, *p < 0.05, *p < 0.01.
Targeted lipidomics was then performed to measure CoQ_6_ levels and intermediates in the CoQ_6_ biosynthetic pathway in coq5Δ harboring empty vector, wild‐type yeast COQ5‐FLAG (WT), or COQ5 ^ G123D ^ ‐FLAG. Western blotting was performed to validate the expression of Coq5‐Flag WT and G123D; Coq5‐Flag G123D was expressed at a slightly lower level compared to WT, indicating that the point mutation could have a small impact on protein stability in addition to catalytic activity (Figure 2B). The coq5Δ strain expressing an empty vector (EV) revealed undetectable CoQ_6_, while the coq5Δ strain with WT was able to fully rescue CoQ_6_ biosynthesis. Interestingly, the coq5 ^ G123D ^ mutation was unable to rescue CoQ_6_ levels (Figure 2C). Correspondingly, the coq5 ^ G123D ^ mutant exhibited a significant increase in the intermediate DDMQ_6_, the substrate of Coq5, as well as the earlier intermediate PPHB_6_, which commonly accumulates when the CoQ pathway is dysfunctional. The result indicates the blockade at the Coq5 reaction step, reflecting a loss‐of‐function impact of the variant p.Gly118Asp on the COQ5 enzyme function.
Discussion
4
Prior to this study, seven individuals from five families have been reported with COQ5‐PCD [9, 10, 11, 12]. Previous reports described patients with a range of clinical manifestations, including retinitis pigmentosa, with or without associated systemic involvement, as well as primarily neurological symptoms, varying in severity. Only one patient has been documented with severe multi‐systemic involvement. Here, we present two additional cases, both exhibiting severe multi‐systemic disease, thereby increasing the total number of reported patients to nine, derived from six families. Among those, most patients presented with developmental delay and intellectual disability (7/9, 77.8%). Seizure and ataxia are also common (62.5% (5/8) and 60% (3/5), respectively; Table 2). Cerebellar atrophy is frequently associated with *COQ5‐*PCD; however, this was not found in our patients, but instead had other brain MRI abnormalities classically associated with primary mitochondrial disorders, such as abnormal signal hyperintensity in basal ganglia and thalami. Most notably, brain imaging in our patients revealed multiple strokes, which, although associated with other mitochondrial disorders, have not been reported in patients with COQ5‐PCD [24].
Ophthalmologic involvement is common among previously reported patients (6/7, 86%). Eye movement disorders were reported in three patients [9], while RP was a major presentation in two patients [10]. However, optic atrophy has not been previously reported. Thus, our study further supports the link between COQ5 variants and RP, expanding the ophthalmologic phenotype in COQ5‐PCD. Unlike other primary CoQ_10_ deficiencies, cardiac and renal involvement have not been reported in COQ5‐PCD.
Though our patients had low CoQ_10_ levels in blood, these are not consistently low in COQ5‐PCD, with 85% of the patients (6/7) having decreased CoQ_10_ levels in plasma, leukocyte, or muscle specimens [9, 11]. This variability may be partly due to differences in methodology or tissue sampling, in addition to phenotypic heterogeneity. This highlights the crucial role genetic testing plays in diagnostic confirmation.
Malicdan et al. described a complex intronic variant in the homozygous state that led to reduced COQ5 mRNA and protein expression, supporting a loss‐of‐function mechanism in the pathogenesis of COQ5‐PCD [9]. Subsequently, one nonsense (p.Tyr311Ter) and two canonical splicing variants (c.682–7>G and c.681 + 1G>A) have been reported. Additionally, two missense variants (p.Arg123Trp, and p.Gly118Ser) have been identified; however, their clinical significance remains uncertain. This is partly due to the limited number of missense variants reported to date and the limited predictive accuracy of in silico tools. Furthermore, no functional studies had been conducted to establish the deleterious effects of these missense variants, which limits the strength of the variant‐disease association.
In our current report, our patients presented with the p.Gly118Asp variant in trans with a frameshift variant. The missense variant was located in an exon‐intron boundary, but RNA analysis revealed that the variant does not affect splicing. In order to study the potentially disruptive effect of this variant on the biochemical activity of COQ5, we employed a yeast system. Given its facultative anaerobe nature and its highly conserved mitochondrial biology, S. cerevisiae has been used in studies of mitochondrial disorders and proven effective for evaluating the impact of variants on CoQ biosynthesis [25, 26]. In this study, we measured yeast CoQ_6_, an equivalent to human CoQ_10_, and intermediates in the CoQ_6_ biosynthetic pathway. This revealed that yeast harboring a variant equivalent to human p.Gly118Asp had decreased CoQ_6_ levels accompanied by an accumulation of the COQ5 yeast‐equivalent substrate, DDMQ_6_. These findings support the loss‐of‐function effect of the p.Gly118Asp variant and further emphasize the use of yeast as a model system for COQ5‐PCD as well as other primary CoQ_10_ deficiencies.
Interestingly, two missense variants affecting the same amino acid residue, p.Gly118Ser and p.Gly118Asp, have been identified in COQ5‐PCD. The patient homozygous for p.Gly118Ser exhibited profound developmental delay, although information regarding systemic involvement was unavailable [12]. A patient with compound heterozygosity for p.Gly118Ser and a splice variant presented with severe multisystemic disease [11]. These observations suggest that glycine at position 118 plays a crucial role in COQ5 function, and variants affecting this residue may be associated with more severe multisystemic COQ5‐PCD. The Gly118 residue is highly conserved and resides within the DVAGGSG segment, which is essential for the binding of S‐adenosylmethionine (SAM), a key methyl donor [27]. Disruption of this segment may impair SAM binding, leading to a significant loss of enzyme function. These findings may suggest a genotype–phenotype correlation in COQ5‐PCD.
CoQ_10_ supplementation is one of the treatment strategies in primary CoQ_10_ deficiencies; however, patients' responses could be variable [6]. Patients with COQ5‐PCD with ataxia as a predominant symptom have shown subjective improvement in ataxia following CoQ_10_ supplementation [9]. Therefore, we supplemented patient 1 with 15 mg/kg/day of CoQ_10_. The parents reported subjective improvement in her social interaction. There was also a suggestion of reduced stroke risk in that surveillance brain MRIs did not show new strokes while the patient was supplemented. It is possible that CoQ_10_ supplementation may be beneficial among patients with COQ5‐PCD, especially among the patients with reduced CoQ_10_ levels. However, given some of the reported patients maintained normal CoQ_10_ levels, it is likely that decreased CoQ_10_ is not the only underlying pathophysiology of COQ5‐PCD, as well as other primary CoQ_10_ deficiencies [10]. Therefore, supplementation of CoQ_10_ may provide benefit only in certain patients. Larger cohorts and a more complete understanding of the disease are needed to provide robust evidence for the benefit of CoQ_10_ supplementation [3].
Here, we described a pair of siblings with COQ5‐PCD and demonstrated the use of a yeast system as a model to study COQ5‐PCD. We also highlighted intrafamilial variability and demonstrated that recurrent stroke and optic atrophy are possible manifestations of COQ5‐PCD. Given the variable degree of previously reported patients ranging from isolated RP to severe multisystemic disorders, larger cohorts of patients are needed to establish neurologic and extra‐neurologic phenotypes, as well as genotype–phenotype correlation. This study expanded the genotypic and phenotypic spectra of the disease.
Author Contributions
P.W. and J.L.G. designed and conceptualized the study. P.W., S.T., J.A.H., and J.L.G. performed clinical analysis of the patients. P.W., J.L.G., and D.J.W. performed variant analysis. D.J.W. performed RNA analysis. R.M.G. and D.J.P. performed functional study in yeast model system. P.W. drafted the manuscript. All authors were involved with revising the manuscript. P.W. and J.L.G. obtained consents. P.W., J.L.G., and D.J.P. supervised the study.
Ethics Statement
The guardian of the patient signed a consent form for publication approved by Washington University IRB (Media Authorization for the Use and Disclosure of Protected Health Information).
Consent
Consent was obtained from the patient's family for publication of this report.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. RNA studies. (A) Splicing prediction in Alamut Software. Variant allele shows a mild decrease in 3′ splice site strength. (B) Primers used to amplify exons 1–4 of COQ5 cDNA. (C) Agarose gel electrophoresis of exons 1–4 of COQ5 cDNA PCR from proband. Lane 1: DNA ladder, lanes 2–3: Proband, lane 4: mom, lane 5: dad. (D) Deep next generation sequencing of cDNA PCR product. No alternative splicing was seen in 2 proband samples nor a control sample, despite sequencing to depths of > 1500 reads. 82% of reads contained the G118D mutant allele, while only 18% of reads contained the GT deletion allele, confirming degradation of the GT deletion allele, likely due to nonsense mediated decay.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1U. C. Tran and C. F. Clarke , “Endogenous Synthesis of Coenzyme Q in Eukaryotes,” Mitochondrion 7, no. Suppl (2007): S 62–S 71.17482885 10.1016/j.mito.2007.03.007PMC 1974887 · doi ↗ · pubmed ↗
- 2J. A. Stefely and D. J. Pagliarini , “Biochemistry of Mitochondrial Coenzyme Q Biosynthesis,” Trends in Biochemical Sciences 42, no. 10 (2017): 824–843.28927698 10.1016/j.tibs.2017.06.008PMC 5731490 · doi ↗ · pubmed ↗
- 3Y. Wang , N. Lilienfeldt , and S. Hekimi , “Understanding Coenzyme Q,” Physiological Reviews 104, no. 4 (2024): 1533–1610.38722242 10.1152/physrev.00040.2023 PMC 11495197 · doi ↗ · pubmed ↗
- 4A. M. Awad , M. C. Bradley , L. Fernandez‐Del‐Rio , A. Nag , H. S. Tsui , and C. F. Clarke , “Coenzyme Q(10) Deficiencies: Pathways in Yeast and Humans,” Essays in Biochemistry 62, no. 3 (2018): 361–376.29980630 10.1042/EBC 20170106 PMC 6056717 · doi ↗ · pubmed ↗
- 5V. Emmanuele , L. C. Lopez , A. Berardo , et al., “Heterogeneity of Coenzyme Q 10 Deficiency: Patient Study and Literature Review,” Archives of Neurology 69, no. 8 (2012): 978–983.22490322 10.1001/archneurol.2012.206PMC 3639472 · doi ↗ · pubmed ↗
- 6L. Salviati , E. Trevisson , C. Agosto , M. Doimo , and P. Navas , “Primary Coenzyme Q(10) Deficiency Overview,” in, ed. M. P. Adam , J. Feldman , G. M. Mirzaa , R. A. Pagon , S. E. Wallace , and A. Amemiya (Gene Reviews((R)), 1993).28125198 · pubmed ↗
- 7D. Mantle , L. Millichap , J. Castro‐Marrero , and I. P. Hargreaves , “Primary Coenzyme Q 10 Deficiency: An Update,” Antioxidants (Basel) 12, no. 8 (2023): 1652.37627647 10.3390/antiox 12081652 PMC 10451954 · doi ↗ · pubmed ↗
- 8T. P. Nguyen , A. Casarin , M. A. Desbats , et al., “Molecular Characterization of the Human COQ 5 C‐Methyltransferase in Coenzyme Q 10 Biosynthesis,” Biochimica et Biophysica Acta 1841, no. 11 (2014): 1628–1638.25152161 10.1016/j.bbalip.2014.08.007PMC 4331671 · doi ↗ · pubmed ↗