Comparative gene expression analysis in closely related dermatophytes reveals secondary metabolism as a candidate driver of virulence
Lenka Machová, Martin Kostovčík, Karel Švec, Vít Hubka, Miroslav Kolařík, Adéla Wennrich

TL;DR
A study on fungal skin pathogens finds that genes involved in making secondary metabolites are more active in a highly infectious strain, possibly explaining its spread.
Contribution
Identifies secondary metabolism as a potential driver of virulence in emerging dermatophyte strains through comparative gene expression analysis.
Findings
Genes related to secondary metabolism are upregulated in T. benhamiae var. luteum under ex vivo skin conditions.
Two biosynthetic gene clusters are linked to known metabolites, while others remain uncharacterized.
Enhanced gene expression may explain increased infectivity and highlights targets for diagnostics or treatments.
Abstract
Dermatophytes are important fungal skin pathogens affecting humans and animals worldwide. Although several virulence factors have been identified using genomic, proteomic, and transcriptomic approaches, their roles remain incompletely understood. In this study, we applied a comparative approach using four closely related taxa within the Trichophyton benhamiae complex, which differ in infectivity despite sharing common hosts. We focused on the emerging zoonotic pathogen T.benhamiae var. luteum, currently responsible for epidemic outbreaks in Europe, and compared it to its less infective relatives. A set of 16 candidate genes, informed by preliminary transcriptomic screening, was assessed via RT-qPCR across 12 strains grown in vitro (Sabouraud dextrose broth) and ex vivo (murine skin explants). Genes associated with secondary metabolism were consistently upregulated under ex vivo…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
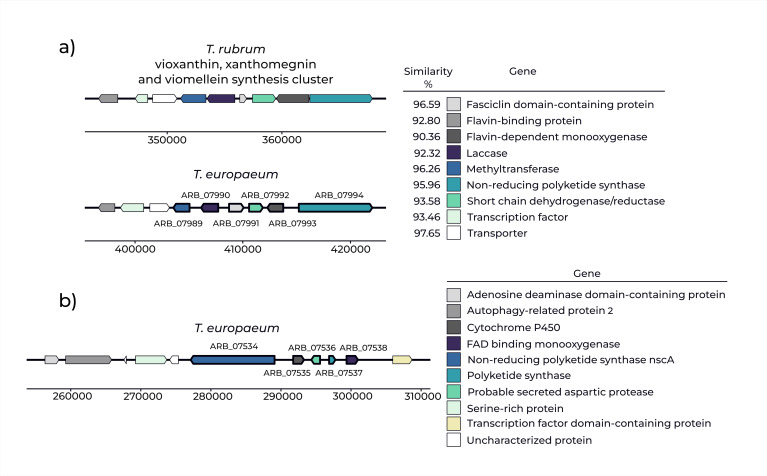 Fig 1
Fig 1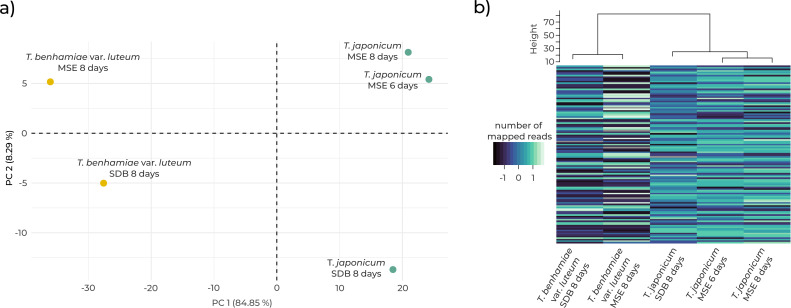 Fig 2
Fig 2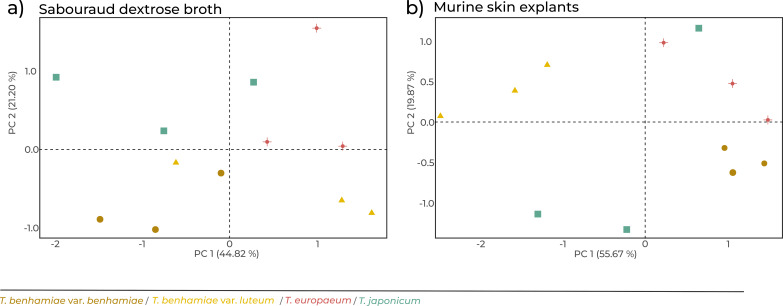 Fig 3
Fig 3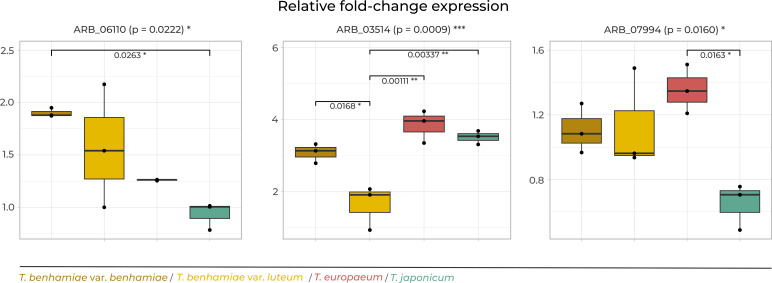 Fig 4
Fig 4 Fig 5
Fig 5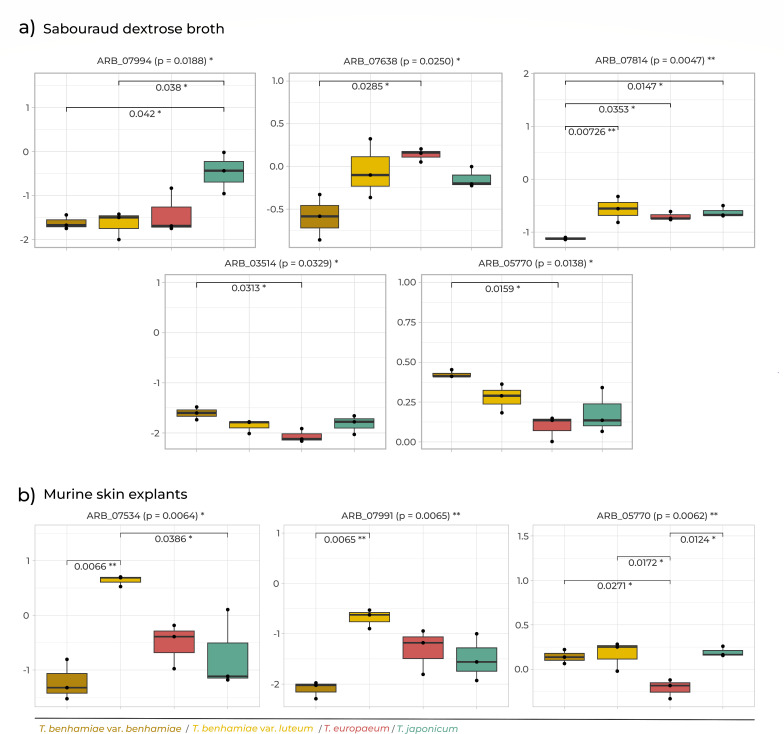 Fig 6
Fig 6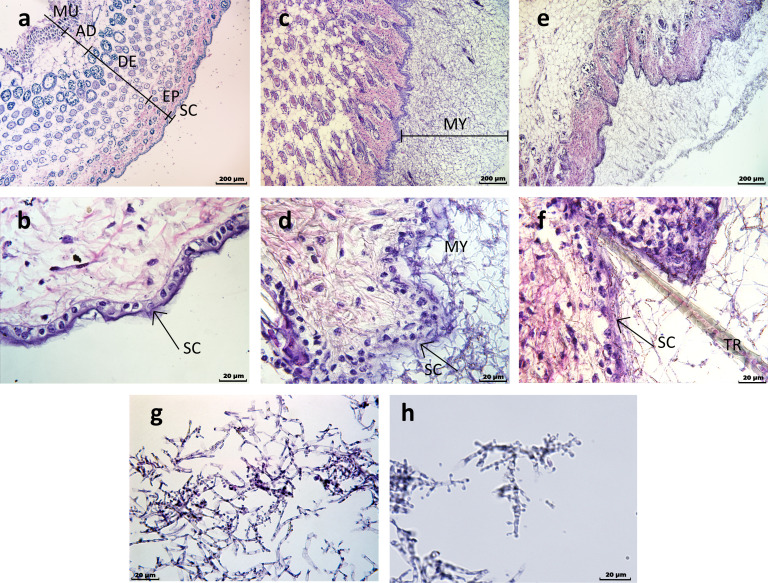 Fig 7
Fig 7| Taxon | Referred to as | Strain ID | CCF identifier | Year of isolation | Source |
|---|---|---|---|---|---|
| a | IHEM 4710 T | CCF6484 | 1956 | Human skin | |
| b | USA 3356 | CCF6486 | 2010 | Dog | |
| c | IHEM 3287 | CCF6483 | 1970 | Monoascosporic isolate no. 3 from crossing of strains IHEM 24908× IHEM 4710 | |
| a | IHEM 25742 | CCF6474 | 2012 | Human skin | |
| b | SK 1248/12 | CCF4852 | 2012 | Skin near mouth of a 6-year-old girl | |
| c | IHEM 25077 | CCF6475 | 2011 | Human scalp and neck skin | |
|
| a | IHEM 20161 = CBS 112371 | CCF6479 | 2002 | Face of human (after contact with guinea pigs) |
| b | IHEM 25062 | CCF6477 | 2011 | Face of human | |
| c | ME 192/12 | CCF6380 | 2012 | Thigh skin of a 16-year-old girl | |
|
| a | IHEM 17701T | CCF6481 | 1963 | Human skin |
| b | VUT 97010 | CCF6489 | 1997 | Rabbit | |
| c | NUBS 13002 | CCF6488 | Unknown | Human skin |
| Locus | Log2 fold | Adjusted | Annotation NCBI | Annotation UniProt | Possible function |
|---|---|---|---|---|---|
| ARB_06975 | −13.80 | 5.27 × 10−12 | Hydrophobin, putative | Hydrophobin A | Surface hydrophobicity, adherence |
| ARB_05303 | 6.26 | 5.84 × 10−08 | Uncharacterized protein | Sulfotransferase family protein | Sulfur-related processes |
| ARB_00847 | −11.16 | 3.45 × 10−07 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_07538 | 6.84 | 2.12 × 10−06 | FAD binding monooxygenase, putative | FAD binding monooxygenase, putative | Secondary metabolism, cluster B |
| ARB_03989 | −10.49 | 1.56 × 10−05 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_04141 | −8.46 | 4.18 × 10−05 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_01369 | −10.71 | 7.33 × 10−05 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_07952 | −8.10 | 0.00011882 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_05947 | 6.72 | 0.00012002 | Uncharacterized protein | FAD-binding PCMH-type domain-containing protein | Undetermined |
|
|
|
|
|
|
|
| ARB_04859 | 5.05 | 0.00033383 | Oxalate decarboxylase, putative | Probable oxalate decarboxylase | Primary metabolism |
| ARB_02085 | −9.14 | 0.00042272 | Uncharacterized protein | Methyltransferase domain-containing protein | Various biochemical reactions |
| ARB_07989 | 7.98 | 0.00054269 | O-methyltransferase, putative | O-methyltransferase, putative | Secondary metabolism, cluster A |
| ARB_07993 | 7.53 | 0.00054269 | Uncharacterized protein | Uncharacterized protein | Secondary metabolism, cluster A |
|
|
|
|
|
|
|
| ARB_07518 | −9.63 | 0.00069182 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_04430 | −8.78 | 0.00069182 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_07536 | 8.80 | 0.00087444 | Uncharacterized protein | Probable secreted aspartic protease | Secondary metabolism, cluster B |
| ARB_04594 | 8.38 | 0.00087444 | Uncharacterized protein | STAS domain-containing protein | Sulfur-related processes |
|
|
|
|
|
|
|
| ARB_07951 | −8.30 | 0.00087739 | Uncharacterized protein | DUF3328 domain protein | Undetermined |
| ARB_06429 | −9.81 | 0.00087739 | O-methyltransferase | O-methyltransferase | Various biochemical reactions |
| ARB_05141 | −8.65 | 0.00087739 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_05392 | −7.73 | 0.00098404 | Uncharacterized protein | Chitinase | Cell wall-related processes |
| ARB_02932 | 6.79 | 0.00133298 | RTA1 domain protein, putative | RTA1 domain protein, putative | Undetermined |
| ARB_07276 | 4.72 | 0.00157637 | Uncharacterized protein | phosphoadenosine phosphosulfate reductase domain-containing protein | Sulfur |
| ARB_05274 | 4.96 | 0.00195658 | Conserved uncharacterized protein | Uncharacterized protein | – |
| ARB_04644 | −5.81 | 0.00208368 | Uncharacterized protein | Lumazine-binding protein | Undetermined |
| ARB_06173 | 4.66 | 0.00208841 | Zinc-containing alcohol dehydrogenase, | Zinc-containing alcohol dehydrogenase, | Undetermined |
| ARB_01072 | 7.31 | 0.00210063 | Carboxylesterase, putative | Carboxylic ester hydrolase | Various biochemical reactions |
| ARB_03721 | 5.78 | 0.00256559 | Uncharacterized protein | AB hydrolase-1 domain-containing protein | Undetermined |
| ARB_07517 | −7.66 | 0.00270752 | Uncharacterized protein | Acyl-protein thioesterase 1 | Sulfur-related processes |
| ARB_07246 | −9.05 | 0.00270752 | Uncharacterized protein | DUF1203 domain protein | Undetermined |
| ARB_01258 | −3.87 | 0.00301641 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_01444 | −7.01 | 0.00329636 | Uncharacterized protein | Glucan endo-1,3-beta-D-glucosidase | Cell wall-related processes |
| ARB_07535 | 7.25 | 0.00386691 | Uncharacterized protein | Cytochrome P450 | Secondary metabolism, cluster B |
| ARB_05140 | −7.40 | 0.00396502 | Uncharacterized protein | Protein kinase domain-containing protein | Basic cellular processes |
| ARB_01724 | −5.24 | 0.0041556 | Hsp90 binding co-chaperone (Sba1), putative | Hsp90 binding co-chaperone (Sba1), putative | Basic cellular processes |
|
|
|
|
|
|
|
| ARB_04955 | −7.43 | 0.0041556 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_01579 | −7.22 | 0.00518502 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_07992 | 6.66 | 0.00613564 | Short chain dehydrogenase/reductase family protein | Short chain dehydrogenase/reductase family protein | Secondary metabolism, cluster A |
|
|
|
|
|
|
|
| ARB_00424 | 3.33 | 0.0069251 | Uncharacterized protein | Glutathione transferase | Sulfur-related processes |
| ARB_05391 | 3.76 | 0.0069251 | PTR family peptide transporter, putative | PTR family peptide transporter, putative | Transmembrane transport |
| ARB_00651 | −7.26 | 0.00707818 | Uncharacterized protein | HNH nuclease domain-containing protein | Undetermined |
| ARB_06241 | −6.27 | 0.00867606 | Opsin, putative | Opsin, putative | Undetermined |
| ARB_05812 | 4.24 | 0.00962227 | Uncharacterized protein | Fatty acid hydroxylase domain-containing | Primary metabolism |
| ARB_03863 | −7.33 | 0.01095632 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_01940 | 4.02 | 0.01095632 | Homogentisate 1,2-dioxygenase, putative | Homogentisate 1,2-dioxygenase | Various biochemical reactions |
| ARB_03395 | −4.26 | 0.01250736 | Solid-state culture expressed protein (Aos23), putative | Solid-state culture expressed protein (Aos23), putative | Undetermined |
| ARB_06838 | −6.83 | 0.01278189 | GPI-anchored endo-1,3(4)-beta-glucanase, | GPI-anchored endo-1,3(4)-beta-glucanase, putative | Cell wall-related processes |
|
|
|
|
|
|
|
| ARB_01913 | −7.08 | 0.01288591 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_02208 | 3.52 | 0.0133612 | Uncharacterized protein | Oxalate decarboxylase | Primary metabolism |
|
|
|
|
|
|
|
| ARB_06800 | −6.79 | 0.01489926 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_07966 | 3.61 | 0.0151062 | Uncharacterized protein | Non-reducing polyketide synthase (NscA) | Secondary metabolism |
| ARB_01650 | −5.56 | 0.0158866 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_00746 | 5.89 | 0.0158866 | Uncharacterized protein | Major facilitator superfamily (MFS) profile domain- | Transmembrane transport |
| ARB_04467 | −3.58 | 0.01673777 | Glucanase, putative | Probable glucan 1,3-beta-glucosidase | Cell wall-related processes |
| ARB_04431 | −6.92 | 0.01749836 | Uncharacterized protein | altered inheritance of mitochondria protein 9, mitochondrial | Undetermined |
| ARB_01248 | 4.09 | 0.02039071 | Zinc-containing alcohol dehydrogenase, | Zinc-containing alcohol dehydrogenase, | Undetermined |
| ARB_00248 | −6.96 | 0.02259337 | NAD-dependent epimerase/dehydratase | NAD-dependent epimerase/dehydratase | Undetermined |
| ARB_01027 | 3.10 | 0.02563679 | Uncharacterized protein | MFS-type efflux pump MFS1 | Transmembrane transport |
| ARB_07899 | −6.84 | 0.02563679 | Folylpolyglutamate synthetase, putative | Folylpolyglutamate synthase | Basic cellular processes |
| ARB_06251 | −7.70 | 0.02846453 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_04058 | −3.95 | 0.02918407 | DNA repair and transcription factor Ada, | DNA repair and transcription factor Ada, | Basic cellular processes |
| ARB_07483 | −3.10 | 0.02966119 | Heat shock protein Awh11, putative | Heat shock protein Awh11, putative | Basic cellular processes |
| ARB_03753 | 5.25 | 0.02971309 | Uncharacterized protein | Major facilitator superfamily (MFS) profile domain- | Transmembrane transport |
| ARB_02282 | −6.74 | 0.03048244 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_06102 | −6.47 | 0.03048244 | Uncharacterized protein | Aminoglycoside phosphotransferase domain- | Various biochemical reactions |
| ARB_04769 | 6.71 | 0.03048244 | Uncharacterized protein | Probable neutral protease 2 homolog | Secreted protease |
| ARB_05177 | −6.92 | 0.03148407 | Uncharacterized protein | Upregulated in Daf-2 domain-containing | Undetermined |
| ARB_03519 | 5.33 | 0.03163056 | Uncharacterized protein | RTA1 domain protein | Undetermined |
| ARB_07381 | −5.84 | 0.03260191 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_03167 | −6.36 | 0.03381736 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_07537 | 6.21 | 0.03381736 | Polyketide synthase, putative | Polyketide synthase, putative | Secondary metabolism, cluster B |
| ARB_06539 | −7.51 | 0.03381736 | Uncharacterized protein | Uncharacterized protein | – |
|
|
|
|
|
|
|
| ARB_06768 | −3.47 | 0.03754576 | Phosphate-repressible phosphate permease, putative | Phosphate transporter | Transmembrane transport |
| ARB_05610 | −6.21 | 0.03754576 | Uncharacterized protein | Uncharacterized protein | – |
| ARB_03617 | −2.59 | 0.03890598 | Uncharacterized protein | LEA domain protein | Undetermined |
| ARB_05078 | −5.59 | 0.04451843 | Uncharacterized protein | Alpha-carbonic anhydrase domain-containing protein | Undetermined |
| Locus | Protein name | Function/activity | Source |
|---|---|---|---|
| ARB_00701 | Subtilisin 3 (Sub3) | Keratinolytic protease ( | ( |
| ARB_06110 | Dipeptidyl peptidase (DPP) IV | Keratinolytic protease ( | ( |
| ARB_06651 | Dipeptidyl peptidase (DPP) V | Keratinolytic protease ( | ( |
| ARB_00494 | Leucin aminopeptidase 2 (Lap 2) | Keratinolytic protease ( | ( |
| ARB_04170 | Aspartic-type endopeptidase (OPSB) | Peptidase ( | ( |
| ARB_07638 | Malate synthase | Glyoxylate cycle ( | ( |
| ARB_07814 | Isocitrate lyase | Glyoxylate cycle ( | ( |
| ARB_03514 | Class III chitinase ChiA2 | Cell wall growth and remodeling ( | RNAseq analysis |
| ARB_05770 | 1,3-Beta-glucanosyltransferase | Cell wall growth and remodeling ( | RNAseq analysis |
| ARB_02741 | GPI-anchored CFEM domain protein | Cell wall stability ( | RNAseq analysis |
| ARB_07818 | Cytochrome P450 monooxygenase | Metabolism ( | RNAseq analysis |
| ARB_04645 | Catalase easC | Ergot alkaloids synthesis ( | RNAseq analysis |
| ARB_07994 | Pigment polyketide synthase from cluster A | Vioxanthin, xanthomegnin, and viomellein synthesis ( | RNAseq analysis |
| ARB_07991 | Fasciclin domain family protein (cluster A) | Vioxanthin, xanthomegnin, and viomellein synthesis ( | RNAseq analysis |
| ARB_07534 | Polyketide synthase from cluster B | Secondary metabolism ( | RNAseq analysis |
| ARB_03861 | Glutathione S-transferase Ure2-like | Stress management ( | RNAseq analysis |
- —Ministry of Educationhttp://dx.doi.org/10.13039/100009122
- —Czech Academy of Scienceshttp://dx.doi.org/10.13039/501100004240
- —Czech Academy of Scienceshttp://dx.doi.org/10.13039/501100004240
- —Czech Academy of Scienceshttp://dx.doi.org/10.13039/501100004240
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNail Diseases and Treatments · Plant Pathogens and Fungal Diseases · Fungal Biology and Applications
INTRODUCTION
Dermatophytes are widespread fungal pathogens with a prevalence of 20%–25% in the world’s human population (1). In recent decades, awareness of these fungi has grown due to the rise in antifungal-resistant strains and the discovery of new species (2–4). One major public health issue is the rapid spread of Trichophyton benhamiae var. luteum, commonly referred to as “yellow-phenotype strains” of T.benhamiae in Europe. This emerging zoonotic pathogen has spread extensively in European guinea pig farms and pet shops, and among their owners and their children, especially young individuals (4–6). Interestingly, closely related species, T.japonicum and T.europaeum (commonly referred to as “white-phenotype strains”), have been present on the same hosts in Europe for decades without triggering similar outbreaks and epidemics (4). Coinfection of guinea pigs with both T.benhamiae var. luteum and “white-phenotype strains” (i.e., T.europaeum and T.japonicum) has been documented; however, the epidemic strains of T.benhamiae var. luteum cause human infections up to 30 times more frequently and up to four times more frequently in guinea pigs than its closely related species (4, 6, 7). One explanation for the lower incidence of T.japonicum and T.europaeum compared with T. benhamiae var. luteum in guinea pig and human populations may be their lower infectivity. In addition, a population of a different variety (T.benhamiae var. benhamiae) belonging to the same species as the epidemic strains (T.benhamiae var. luteum) occurs in North America, yet no reports of epidemics caused by these strains have been documented in that area (4). It remains unclear why the epidemic strain spreads so effectively among European guinea pigs and children compared with its close relatives or even other populations of the same species.
Dermatophytes have developed various mechanisms for spreading within hosts and invading their tissues. A key feature is their sophisticated machinery for degrading keratin and other skin components, primarily through the secretion of proteases. Notable examples include subtilisin-like proteases, dipeptidyl peptidases, leucine aminopeptidases, and metallopeptidases (8, 9). Hosts counteract dermatophyte infections using several defense mechanisms, such as high body temperature, and the mildly acidic pH of the skin, as well as innate immunity like keratinocytes, macrophages, neutrophils, and adaptive immunity, especially the Th1 and Th17 cell-mediated response (10, 11). Dermatophytes can evade some host barriers and produce a wide range of proteases that vary according to the environmental pH (12–15). As a result of keratin degradation by dermatophytes, the pH of the surrounding area increases, which impairs the host’s natural immunity (14, 16). Compounds produced by dermatophytes, such as penicillin G, melanin, xanthomegnin, and vioxanthin are known or suspected toxins and/or immunomodulators (17–20). Nevertheless, the role of secondary metabolites in dermatophyte pathogenesis remains underexplored.
Despite the high genetic similarity among dermatophytes (4, 21), different species exhibit distinct host preferences and varying levels of virulence across host species. These differences likely arise from variations in gene expression and enzyme activity regulation (22). Several approaches can be used to identify potential virulence factors. In dermatophytes, comparative studies have primarily focused on gene expression during in vitro saprobic and in vivo parasitic growth phases (23, 24). Various alternatives to the in vivo approach have been introduced as useful, more ethical, cost-effective, and reproducible options (25). Several ex vivo skin models have been developed in addition to the traditional in vitro models to better replicate real conditions of infection in dermatophytes (26–31). These include reconstructed human epidermis (30) and organoid-based models (31), which offer human tissue relevance but may lack full skin architecture, immune elements, or mechanical barrier integrity. In contrast, animal-derived explants (26–29) preserve native skin structure and allow short-term fungal colonization under near-physiological conditions. Another option is the comparison of related species differing in infectivity or virulence. However, this requires the use of very closely related populations or species to ensure that the observed results are not simply attributed to differences between species during their speciation. This comparative approach has not been used for dermatophytes.
In this study, we applied a comparative gene expression approach to identify candidate virulence-associated genes in dermatophytes. We compared more infective taxa with less infective, yet closely related ones to identify differences in gene expression potentially linked to virulence. This approach allows us to distinguish general virulence factors shared across taxa from those potentially associated with enhanced infectivity. Candidate genes were selected based on preliminary transcriptomic data and literature evidence, and their expression was then validated across a broader strain data set using RT-qPCR to strengthen the robustness of observed patterns.
MATERIALS AND METHODS
Strains and cultivation conditions
A total of 12 strains—three from each of the four closely related taxa, T.benhamiae var. benhamiae, T.benhamiae var. luteum, T.japonicum, and *T.europaeum—*were used (Table 1). All strains are publicly available from the Culture Collection of Fungi, Charles University (CCF). Species identification was confirmed using ITS rDNA sequencing (4). Cultures were maintained on malt extract agar (MEA; HiMedia) at 6 °C in the dark. Prior to the experiment, all strains were subcultured to ensure purity. To minimize the risk of contamination, a single-colony isolation step was performed by spreading diluted inoculum on MEA plates and selecting a single morphologically typical colony for further use. Strains were also briefly cultured in liquid medium supplemented with chloramphenicol, and bacterial contamination was ruled out by microscopic inspection.
To prepare the inoculum, strains were grown in Sabouraud dextrose broth (SDB; HiMedia) at 30 °C for 8 days, shaking at 200 rpm (Digital Orbital Shaker, Heathrow Scientific). The resulting biomass was homogenized by vortexing and diluted to obtain approximately equal cell densities. This method was used instead of standard spore suspensions because some strains—particularly T.benhamiae var. luteum—were sterile.
Three to five biological replicates were prepared for each strain and for both cultivation conditions: liquid culture and ex vivo skin model. Liquid cultures were initiated by inoculating 30 mL of SDB with 4 µL of homogenate and incubated under the same conditions described above.
Murine skin models
The ex vivo murine skin explant (MSE) model protocol was developed and optimized based on published methodologies (31–33). Healthy three-month-old male BALB/c mice were obtained from the breeding facility of the Institute of Microbiology, CAS. All procedures involving animals were conducted in full compliance with Czech legislation (Act No. 246/1992 Coll., on the protection of animals against cruelty) and the Guidelines for the Care and Use of Laboratory Animals.
The dorsal skin of live mice was shaved using an electric clipper (Aesculap Exacta GT 416; B. Braun, Germany), and the animals were subsequently euthanized. The shaved area was swabbed with 70% ethanol. Skin sections (2 × 2 cm) were excised under sterile conditions, cleaned of adipose tissue, and placed into 0.1% benzalkonium bromide solution for disinfection.
Within 30 minutes, samples were washed with ice-cold phosphate-buffered saline (PBS; Sigma-Aldrich), immersed in 1% Penicillin-Streptomycin (Pen-Strep; Sigma-Aldrich) for 10 minutes, then washed twice again with PBS and Pen-Strep. Finally, skin samples were placed on sterile gauze in 6-well plates.
Each sample received 1 mL of murine skin fluid (MSF), composed of 10% (v/v) heat-inactivated fetal bovine serum (Gibco), 8% (v/v) MSF buffer, and 1% (v/v) Pen-Strep. MSF buffer (adjusted to pH 6.4) contained: 8.0 g/l NaCl, 0.4 g/l KCl, 0.0875 g/l Na₂HPO₄, 0.0625 g/l KH₂PO₄, 0.2 g/l MgSO₄, and 6.25 g/l D-glucose.
Four microliters of SDB culture supernatant, microscopically confirmed to be free of bacterial contamination, was added to each skin sample. The samples were incubated at 30 °C and 5% CO₂ for 8 days without shaking (MCO-170AICUV-PE incubator, Panasonic). MSF was refreshed daily. The 8-day cultivation period was chosen based on a pilot RNAseq analysis.
RNA extraction and processing
Mycelia from liquid cultures were harvested by centrifugation at 1000 rpm for 10 minutes. Mycelium or colonized skin was ground in liquid nitrogen, resuspended in 1 mL of TRIzol reagent (Thermo Fisher Scientific), and stored at –35 °C until RNA isolation. RNA was extracted following the manufacturer’s instructions (34). Equal amounts of total RNA were used for downstream cDNA synthesis, but fungal biomass was not quantified separately from host tissue in ex vivo samples.
RNA concentration and purity were assessed using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). Genomic DNA was removed with the TURBO DNA-free Kit and protected using RNaseOUT Recombinant Ribonuclease Inhibitor (both Thermo Fisher Scientific). RNA integrity was confirmed by 1% agarose gel electrophoresis stained with ethidium bromide (HiMedia). Concentration was measured using Qubit RNA HS and BR Assay Kits on a Qubit 2.0 Fluorometer (Thermo Fisher Scientific).
Candidate gene identification by RNA sequencing
To identify genes potentially involved in virulence and host interaction, RNA-seq was performed on two representative strains—T.benhamiae var. luteum (IHEM 25742) and T.japonicum (NUBS 13002)— under both in vitro and ex vivo conditions.
RNA quality was further verified using the Agilent 2100 Bioanalyzer and the RNA 6000 Nano Kit (Agilent Technologies). Due to moderate RNA degradation (RIN <8), library preparation was adjusted accordingly.
Ribosomal RNA was depleted using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs), and libraries were prepared with the KAPA RNA HyperPrep Kit (Roche). Twenty nanograms of RNA was used for input; fragmentation was performed at 85 °C for 2 minutes, followed by 14 amplification cycles. Indexed adapters (KAPA Dual-Indexed Adapter Kit, Roche) were used, and sequencing was performed on the Illumina NextSeq 500 platform.1
Reads were aligned using STAR v.2.7.1 (35) and mapped with Bowtie2 (36) to a genome index based on T.europaeum strain CBS 112371 (RefSeq: GCF_000151125.1). Differential expression analysis was conducted in R v.3.6.0 using the DESeq2 package (37), with standard error and log₂ fold change computed. The Wald test and Benjamini-Hochberg correction were applied. All replicates from both conditions were used. Genes with adjusted P-values < 0.05 were considered significant and annotated using NCBI and UniProt databases.
RT-qPCR analysis
For RT-qPCR, 1 µg of total RNA was reverse-transcribed using the LunaScript RT SuperMix Kit (New England Biolabs) according to the manufacturer’s protocol. Complementary DNA (cDNA) from three high-quality biological replicates was pooled in equal amounts prior to qPCR. This approach was chosen to minimize noise from individual-level variation and to ensure sufficient cDNA quantity across the large panel of target genes and conditions. We acknowledge that pooling limits the ability to assess within-strain variability.
Quantitative PCR was performed using the CFX96 Real-Time System (Bio-Rad Laboratories) and the Luna Universal qPCR Master Mix (New England Biolabs). The PCR reaction was done with 0.5 µL of 10 pM primers, and 0.5 µL of cDNA in the final volume of 20 µL. The reaction comprised an initial denaturation at 95 °C for 60 s, followed by 39 cycles of 95 °C for 15 s and 60 °C for 30 s, inactivation at 95 °C for 60 s, cooldown at 54 °C for 60 s, and final cooldown at 25 °C for 60 s. Melting curve analysis was conducted by increasing the temperature from 54 °C to 95 °C in 0.5 °C increments every 10 s. Each measurement was performed in duplicate, and if the standard deviation exceeded 2, the experiment was repeated.
Primer design and efficiency testing
Primer pairs for ARB_00701 and TERG_04919 were adapted from previously published studies (23, 38). All other primers were designed using Primer-BLAST (NCBI) based on the genome sequence of T.europaeum CBS 112371 (RefSeq: GCF_000151125.1). Each primer pair was designed such that at least one primer spanned an exon–exon junction to avoid amplification of genomic DNA.
The OligoAnalyzer tool (IDT) was used to assess potential secondary structures, with accepted primers showing ΔG > –7 J for dimers and > –4 J for hairpins. All primer pairs were tested using pooled cDNA and serial dilutions (1:10; 1:50; 1:250; 1:1,250; 1:6,250). Primer efficiencies ranged from 85% to 100% (Supplemental material 2a).
RT-qPCR data processing and analysis
The stability and reliability of four previously published reference genes were analyzed using RefFinder (39), which integrates the tools BestKeeper (40), NormFinder (41), and geNorm (42). Specifically, the analyzed genes were ARB_06116 (DNA-directed RNA polymerase II subunit RPB2*; rpb2*) (43), ARB_03667 (ADP-ribosylation factor; adp-rf) (44, 45), ARB_00512 (Mitotic cohesin complex subunit Psm1; psm1) (44, 45), and TERG_04919 (Chitin synthase; CHS) (46). Based on this analysis, rpb2 and adp-rf were selected as the most stable reference genes (Supplemental material 2b).
For processing of the acquired RT-qPCR data, the software Bio-Rad CFX Manager v. 3.1 (Bio-Rad Laboratories) was used. Due to the large number of genes examined and the inability to measure the expression of all genes on a single plate for both conditions, the efficiency-weighted method (47) was chosen for data normalization. For the same reason, both reference genes were always run on each plate for all measured samples as an interplate calibrator. The 2^-ΔΔCt^ analysis was performed. As technical replicates were used and cDNA was pooled prior to qPCR, statistical comparisons reflect between-strain variation rather than intra-strain variability. The cycle threshold (Ct) range, response efficiency, coefficient of determination, relative gene expression (ΔCt) values for each strain under different culture conditions, and the effect size measure values (2^-ΔΔCt^) for individual genes are provided in Supplemental material 3. The normal distribution of data was tested with the use of the Shapiro-Wilk test. Based on the results, either ANOVA or Kruskal-Wallis tests were performed, followed by pairwise t-tests. Results are shown in Supplemental material 4.
For the statistical analysis, R v. 4.2.1 (48) was used with the package stats, reshape2 (49), FactoMineR (50), and factoextra (51). The visualization of data was performed with the use of packages from the Tidyverse (52), especially ggplot2 (53), and the packages viridis (54), gridExtra (55), and cowplot (56).
Bioinformatic identification of Biosynthetic Gene Clusters
Biosynthetic gene clusters (BGCs) in the genome of T.benhamiae were identified using the fungal version of antiSMASH v.7.1.0 (57). To assess the level of similarity of genes or clusters of interest, the Ident and Sim tool (https://www.bioinformatics.org/sms2/ident_sim.htmL), BLAST v. 2.15.0 (58), and MAFFT (59) were used. The visualization of clusters was performed in R v. 4.2.1 (48) with the gggenes package (60).
Histological analysis
Prior to RNA extraction, 5 × 5 mm sections of selected skin samples were fixed, embedded, and stained. Fixation was performed in 4% paraformaldehyde and 10% formalin for 48 h (61). Samples were dehydrated using a Leica ASP200s and embedded in paraffin using a Leica EG110 system.
Sections (3 µm) were cut using a Leica RM2125 RTS rotary microtome and placed on precoated slides (Electron Microscopy Sciences). Staining was performed using the standard hematoxylin and eosin (H&E) protocol (62–64), with an additional benzene clearing step. Samples were mounted in Canadian balsam, covered with glass coverslips, and left to harden overnight. H&E-stained sections were used to qualitatively confirm fungal invasion and tissue colonization. No quantitative fungal burden measurement (e.g., by ITS copy number or CFU counts) was performed.
RESULTS
Selection of target genes
A total of 85 genes were significantly differentially expressed (adjusted P < 0.05) between T.benhamiae var. luteum IHEM 25742 and T.japonicum NUBS 13002. The putative functions of more than half were inferred from the predicted protein structure. According to antiSMASH analysis, 11 genes belonged to two biosynthetic gene clusters (BGCs). One cluster was identified as the vioxanthin, xanthomegnin, and viomellein BGC and is referred to here as cluster A; the second, of unknown function, is referred to as cluster B (Table 2; Fig. 1). Additionally, two other genes (ARB_04645, ARB_07966) were identified in the data set (Table 2). ARB_04645 is a component of a previously described ergot alkaloid synthesis pathway in Arthrodermataceae (65). The cluster associated with ARB_07966 could not be identified. Other differentially expressed genes were associated with transmembrane transport, basic cellular functions, cell wall remodeling, and sulfur metabolism (see Supplemental material 1 for the full list of differentially expressed genes).
Biosynthetic gene clusters (BGCs) related to secondary metabolism as predicted by antiSMASH analysis. (a) Cluster A: biosynthetic gene cluster responsible for the synthesis of vioxanthin, xanthomegnin, and viomellein in Trichophyton rubrum (strain CBS 118892), compared with the genome of T.europaeum (strain CBS 112371); similarity values are based on BLAST analysis. (b) Cluster B: uncharacterized biosynthetic gene cluster in T.europaeum with an unknown product. Genes that showed significant differential expression in RNA-seq analysis are annotated and highlighted with bold outlines.
Based on the expression profiles of the 85 significant genes, the samples were divided into two primary clusters that reflected strain identity (Fig. 2a). The first principal component (PC1) accounted for 85% of total variability, separating samples by species, while the second component (PC2) distinguished samples based on cultivation condition. A hierarchical clustering analysis also grouped the samples primarily by species (Fig. 2b). Samples of T.japonicum (NUBS 13002) collected at 6 and 8 days post-inoculation showed minimal variation in gene expression (Fig. 2).
Transcriptomic analysis of epidemic (T.benhamiae var. luteum) and non-epidemic (T.japonicum) strains within the T.benhamiae complex. The figure shows the 85 most significantly differentially expressed genes identified from transcriptomic data. Strains were cultivated in vitro in Sabouraud dextrose broth (SDB) and ex vivo on murine skin explants (MSE). (a) Principal component analysis (PCA) of gene expression variation. (b) Ward’s hierarchical clustering based on expression similarity. Cultivation was terminated at 6 or 8 days post-inoculation.
From these data, 16 genes representing diverse cellular functions were selected for further investigation across a larger data set of 12 strains from the T.benhamiae complex using RT-qPCR on cDNA pooled from biological replicates (Table 3). Nine genes were selected from the transcriptomic data set—seven of which showed significant inter-strain differences, and five were associated with BGCs. Two additional genes (ARB_02741 and ARB_05770) were included due to their presumed functional relevance despite non-significant p-values. Another seven genes were selected based on a literature review as known dermatophyte virulence factors.
Functional annotation of selected genes
Bioinformatic analysis revealed that six genes from cluster A (ARB_07989–ARB_07994) share strong similarity with a laccase-containing vioxanthin, xanthomegnin, and viomellein BGC from T. rubrum (77, 78) (Fig. 1; Supplemental material 5). The five genes in cluster B (ARB_07534–ARB_07538) could not be linked to any known metabolite. The core gene (ARB_07534) was annotated as either a LovB-like polyketide synthase or a non-reducing polyketide synthase encoded by nscA. However, protein-level identity was only 21% with the lovastatin synthase LovB from A. terreus (ATEG_09961) (80) and 17.64% with the neosartoricin synthase NscA from T. tonsurans (TESG_06702) (81). Based on this low sequence similarity, it is unlikely that ARB_07534 performs the same biosynthetic function as either of these enzymes.
RT-qPCR analysis
Gene expression under in vitro and ex vivo conditions
In both cultivation systems, sample clustering reflected species-level differences, except in a few cases: T.benhamiae var. benhamiae vs. var. luteum under in vitro (SDB) conditions, and T.japonicum under ex vivo (MSE) conditions. In the ex vivo model, the species were more clearly separated, and even the two closely related T.benhamiae varieties were distinctly grouped (Fig. 3).
Principal component analysis (PCA) of T.benhamiae complex taxa based on ΔCt values from RT-qPCR data. Samples were obtained from in vitro SDB (a) and ex vivo MSE (b) cultivation.
Gene expression was generally higher under ex vivo conditions, but only three genes showed statistically significant differences (Supplemental material 3c and 4). The DPPIV protease gene (ARB_06110) showed significantly different expression between T.benhamiae var. benhamiae and T.japonicum. T.europaeum significantly overexpressed polyketide synthase ARB_07994 (cluster A) compared with T.japonicum. Additionally, expression of class III chitinase chiA2 (ARB_03514) was significantly lower in T.benhamiae var. luteum than in the other taxa (Supplemental material 4; Fig. 4).
*Relative gene expression fold changes (2-ΔΔCt) for genes with significant differential expression between in vitro (SDB) and ex vivo (MSE). Asterisks indicate statistical significance (*P < 0.05; **P < 0.01; **P < 0.001).
Interestingly, several genes were consistently upregulated under ex vivo conditions across nearly all strains of the T.benhamiae complex. Notably, SUB3 (ARB_00701) with up to sevenfold change, LAP2 (ARB_00494) with up to fivefold change, Class III chitinase chiA2 (ARB_03514) with up to fourfold change, cytochrome P450 monooxygenase (ARB_07818) with up to threefold change, and catalase easC (ARB_04645) with up to fivefold change (Fig. 5; Supplemental material 3c).
Heatmap of relative gene expression fold changes (2-ΔΔCt) for all measured genes under ex vivo (MSE) versus in vitro (SDB) conditions. Includes both statistically significant and non-significant genes.
Differential expression under in vitro conditions
Under in vitro conditions (SDB), moderate differences in gene expression were observed, primarily in genes related to carbohydrate metabolism and cell wall remodeling. These genes include enzymes of the glyoxylate cycle—malate synthase (ARB_07638) and isocitrate lyase (ARB_07814), and cell wall synthesis associated enzyme 1,3-beta-glucanosyltransferase (ARB_05770) and class III chitinase chiA2 (ARB_03514). In all cases, T.benhamiae var. benhamiae formed a distinct cluster from the other taxa. Additionally, T.japonicum showed unique expression of polyketide synthase ARB_07994 from cluster A (Fig. 6a).
*Expression of selected genes that differed significantly between taxa of the T. benhamiae complex when cultivated: (a) in vitro (SDB) and (b) ex vivo (MSE). Asterisks indicate statistical significance (*P < 0.05; **P < 0.01; **P < 0.001).
Differential expression under ex vivo conditions
Under ex vivo conditions on MSE, distinct expression profiles were observed among taxa, especially for genes involved in secondary metabolism. Epidemic strains of T.benhamiae var. luteum showed differential expression of a Fasciclin domain protein (ARB_07991, cluster A) and polyketide synthase ARB_07534 (cluster B). Furthermore, T.europaeum displayed significantly lower expression of 1,3-β-glucanosyltransferase (ARB_05770) compared with other taxa (Fig. 6b).
Evaluation of the ex vivo skin model
To assess the colonization performance of the strains in the ex vivo model, histological analysis of skin samples was performed. Microscopic examination revealed extensive fungal colonization and disruption of the stratum corneum (Fig. 7c through f). Notably, T.benhamiae var. luteum produced microconidia in skin tissue, despite showing limited sporulation on agar media (Fig. 7g and h).
Light microscopy of murine skin explants. (a and b) Control (non-inoculated) skin; (c and d) skin colonized by T.benhamiae var. luteum (IHEM 25077); (e and f) skin colonized by T.japonicum (VUT 97010). (g and h) Sporulation observed on the upper layers of mycelium in IHEM 25077 and VUT 97010, likely as a surface-associated response during extended colonization. Skin layers are labeled: SC, stratum corneum; EP, epidermis; DE, dermis; AD, adipose tissue; MU, muscle; TR, trichome. Mycelial layer is marked as MY.
DISCUSSION
In this study, we used a comparative model of four closely related taxa within the Trichophyton benhamiae complex (T.europaeum, T.japonicum, T.benhamiae var. luteum, and T.benhamiae var. benhamiae). By examining the differences between these closely related taxa, we aimed to identify the factors influencing their varying abilities to adapt and spread in the host population. Similar comparative transcriptomic approaches have been applied to other fungal pathogens to reveal virulence-associated traits (82, 83). However, studies on dermatophytes have largely focused on comparisons among phylogenetically distant species (84). The use of closely related taxa in our model allowed us to detect subtle but biologically significant differences in gene regulation and adaptation.
A key finding of this study was the upregulation of genes involved in secondary metabolism, particularly in the epidemic taxon T.benhamiae var. luteum. Among the candidate genes identified, several showed consistent differential expression (Table 2), with most belonging to two biosynthetic gene clusters: cluster A, which includes the vioxanthin, xanthomegnin, and viomellein biosynthetic genes (represented by genes ARB_07994 and ARB_07991), and cluster B, an uncharacterized cluster (represented by gene ARB_07534) with no close homologs. These results, based on RT-qPCR analysis across multiple strains, suggest that secondary metabolite production may contribute to dermatophyte virulence and host adaptation.
The differential expression patterns were not limited to these clusters. The catalase easC gene (ARB_04645), which is involved in chanoclavine I biosynthesis, a key precursor in the ergot alkaloid pathway (85, 86), also showed substantial variation in expression across strains, suggesting strain-specific regulation of secondary metabolism. Similarly, the polyketide synthase from cluster A (ARB_07994) was significantly underexpressed in T.japonicum under ex vivo conditions (Fig. 4), suggesting environmentally regulated control of polyketide biosynthesis.
Dermatophyte genomes encode more BGCs than other pathogenic fungi (67), yet few have been functionally characterized. Several of their known products—such as vioxanthin, xanthomegnin, and neosartoricin B—are known to have cytotoxic or immunomodulatory effects (18, 86–88). Previous work (89) has shown that secondary metabolite profiles correlate with phylogeny in dermatophytes, indicating that secondary metabolism is under strong evolutionary pressure. The same study identified a distinct group of unique secondary metabolites, particularly sulfur-containing compounds, produced by members of the T.benhamiae complex. Our transcriptomic data further support this view, particularly in the case of sulfur-associated metabolism, where multiple genes showed taxon-specific expression (Table 2). These findings suggest that secondary metabolism may shape ecological adaptation and host-pathogen interactions in the T.benhamiae complex.
In addition to secondary metabolism, we assessed expression of genes encoding secreted proteases, known virulence factors in dermatophytes. In agreement with earlier findings (23, 24), genes, such as SUB3 (ARB_00701), DPPIV (ARB_06110), DPPV (ARB_06651), and LAP2 (ARB_00494), were upregulated under ex vivo conditions. However, their expression was largely uniform across taxa, except for DPPIV, which showed some variation. This suggests that secreted proteases may be core virulence determinants, but not discriminatory markers of virulence variation among closely related taxa.
We also observed upregulation of genes involved in carbohydrate and fatty acid metabolism, such as isocitrate lyase (ARB_07814), under ex vivo conditions. These genes likely reflect adaptation to host-derived substrates, such as keratin and lipids, which are absent in synthetic media.
Gene expression differences were more pronounced under ex vivo conditions than in vitro, reinforcing the biological relevance of the model. Strains grown ex vivo clustered by taxon (Fig. 3), likely reflecting stronger selective pressures in host-mimicking conditions. This underscores the adaptability of dermatophytes in complex environments, where metabolic flexibility is essential for survival. These transcriptional responses likely reflect underlying host–fungus interactions within the skin tissue, which we were only able to assess at a basic level using histological staining. While H&E staining confirmed fungal invasion into skin tissue, it does not provide detailed resolution of fungal–host interactions at the cellular or ultrastructural level. Future studies could benefit from using complementary imaging approaches, such as fluorescent or electron microscopy, to better characterize the spatial dynamics of colonization. Although the ex vivo model is limited by the absence of an active immune response and the use of murine rather than human tissue, it effectively recapitulated differential gene expression and provided a host-mimicking context for assessing fungal adaptation. In contrast, gene expression patterns were highly similar across taxa under in vitro conditions, suggesting that species-specific responses are triggered by environmental factors present only ex vivo, such as the structural complexity of skin and its derivatives.
An interesting deviation from expected clustering patterns was observed in strain ME 192/12, which grouped with T.japonicum despite being genetically assigned to T.europaeum. This may reflect taxonomic ambiguity or gene flow between these closely related species (4). Moreover, expression of cell wall remodeling genes, including chiA2 (ARB_03514) and 1,3-β-glucanosyltransferase (ARB_05770), showed taxon-specific differences. These enzymes play a critical role in fungal cell wall restructuring, potentially contributing to differences in growth morphology and environmental adaptation (90).
Our study provides new insights into the potential virulence factors within the T.benhamiae complex. While secreted proteases are traditionally viewed as major contributors to virulence, their expression showed little variation among taxa. In contrast, secondary metabolism genes displayed notable differential expression, particularly under ex vivo conditions, underscoring the potential role of secondary metabolites in dermatophyte adaptation and pathogenicity. The ability of these fungi to produce unique compounds, including sulfur-containing metabolites, suggests that secondary metabolic pathways are not only shaped by strong evolutionary pressure but may also be essential for host adaptation and survival.
While our data show consistent differential expression of candidate genes, we acknowledge that variation in gene expression under ex vivo conditions may partly reflect differences in fungal biomass among strains. However, the epidemic taxon T.benhamiae var. luteum, which showed the highest expression of several genes, is known to grow more slowly than the other taxa tested. As a result, it likely produced the lowest fungal biomass in the ex vivo model. This supports the interpretation that the observed overexpression reflects true upregulation rather than biomass-driven effects. Furthermore, the use of two validated, stably expressed housekeeping genes (rpb2 and adp-rf) as internal controls for normalization helped reduce bias due to variable fungal loads. Nonetheless, future studies incorporating quantitative fungal burden measurements would help disentangle biomass effects from transcriptional regulation.
Future work should focus on functional validation of genes such as ARB_07534, along with broader comparative studies to better characterize the biosynthetic capacity of dermatophytes. This may include LC-MS-based metabolomic profiling of secondary metabolites or targeted gene disruption (e.g., CRISPR-Cas9 knockout of ARB_07534) to confirm their role in virulence and adaptation. Such research will be essential for advancing targeted diagnostics, antifungal strategies, and our understanding of dermatophyte pathogenesis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Havlickova B, Czaika VA, Friedrich M. 2008. Epidemiological trends in skin mycoses worldwide. Mycoses 51 Suppl 4:2–15. doi:10.1111/j.1439-0507.2008.01606.x 18783559 · doi ↗ · pubmed ↗
- 2Shen JJ, Arendrup MC, Verma S, Saunte DML. 2022. The emerging terbinafine-resistant Trichophyton epidemic: what is the role of antifungal susceptibility testing? Dermatology 238:60–79. doi:10.1159/00051529034058736 · doi ↗ · pubmed ↗
- 3Čmoková A, Rezaei-Matehkolaei A, Kuklová I, Kolařík M, Shamsizadeh F, Ansari S, Gharaghani M, Miňovská V, Najafzadeh MJ, Nouripour-Sisakht S, Yaguchi T, Zomorodian K, Zarrinfar H, Hubka V. 2021. Discovery of new Trichophyton members, T. persicum and T. spiraliforme spp. nov., as a cause of highly inflammatory tinea cases in Iran and Czechia. Microbiol Spectr 9:e 0028421. doi:10.1128/Spectrum.00284-2134468188 PMC 8557871 · doi ↗ · pubmed ↗
- 4Čmoková A, Kolařík M, Dobiáš R, Hoyer LL, Janouškovcová H, Kano R, Kuklová I, Lysková P, Machová L, Maier T, Mallátová N, Man M, Mencl K, Nenoff P, Peano A, Prausová H, Stubbe D, Uhrlaß S, Větrovský T, Wiegand C, Hubka V. 2020. Resolving the taxonomy of emerging zoonotic pathogens in the Trichophyton benhamiae complex. Fungal Divers 104:333–387. doi:10.1007/s 13225-020-00465-3 · doi ↗
- 5Nenoff P, Uhrlaß S, Krüger C, Erhard M, Hipler UC, Seyfarth F, Herrmann J, Wetzig T, Schroedl W, Gräser Y. 2014. Trichophyton species of Arthroderma benhamiae - a new infectious agent in dermatology. J Dtsch Dermatol Ges 12:571–581. doi:10.1111/ddg.1239024981469 · doi ↗ · pubmed ↗
- 6Bartosch T, Frank A, Günther C, Uhrlaß S, Heydel T, Nenoff P, Baums CG, Schrödl W. 2019. Trichophyton benhamiae and T. mentagrophytes target guinea pigs in a mixed small animal stock. Med Mycol Case Rep 23:37–42. doi:10.1016/j.mmcr.2018.11.00530560049 PMC 6290094 · doi ↗ · pubmed ↗
- 7Berlin M, Kupsch C, Ritter L, Stoelcker B, Heusinger A, Gräser Y. 2020. German-wide analysis of the prevalence and the propagation factors of the zoonotic dermatophyte Trichophyton benhamiae. Jo F 6:161. doi:10.3390/jof 603016132899171 PMC 7558194 · doi ↗ · pubmed ↗
- 8Monod M. 2008. Secreted proteases from dermatophytes. Mycopathologia 166:285–294. doi:10.1007/s 11046-008-9105-418478360 · doi ↗ · pubmed ↗