CRISPR/Cas9-compatible plasmids enabling seven dominant genetic selection methods for the human fungal pathogen Cryptococcus neoformans
Michael J. Boucher, Hiten D. Madhani

TL;DR
This paper introduces new genetic selection tools for studying the fungus Cryptococcus neoformans, enabling more efficient genetic experiments.
Contribution
The study introduces blasticidin S resistance as a new selection method and validates phleomycin resistance for genetic manipulation in C. neoformans.
Findings
Blasticidin S resistance via BSD or BSR is a novel dominant selection method for C. neoformans.
Phleomycin resistance via BLE is validated as an additional selection method.
A vector series for CRISPR/Cas9-mediated genome modification was developed and deposited at Addgene.
Abstract
Cryptococcus neoformans is the most common cause of human fungal meningitis and an important model system for studying fundamental eukaryotic biology. Genetic manipulation of this organism relies on three dominant drug resistance markers (nourseothricin acetyltransferase [NAT], neomycin phosphotransferase II [NEO], and hygromycin B phosphotransferase [HYG]) and the recyclable dominant prototrophic marker amdS. With ongoing technological advances that are expanding our ability to explore cryptococcal gene function, contemporary studies often require multiple genetic manipulations in the same strain. Additional dominant selection methods would maximize the utility of these tools by facilitating their combinatorial use. Here, we identify blasticidin S resistance via the blasticidin S deaminase (BSD) or blasticidin S resistance (BSR) markers as a novel dominant selection method for C.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
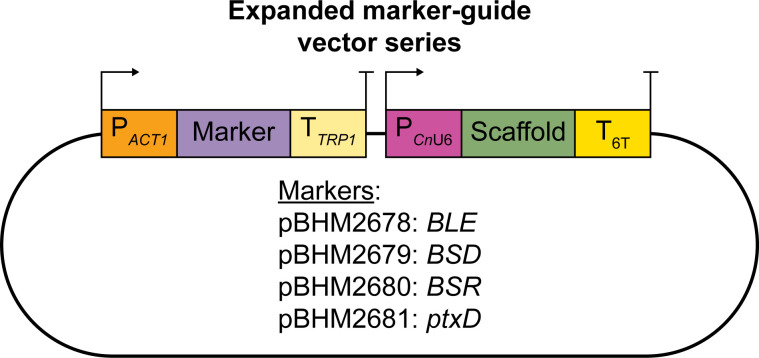 Fig 1
Fig 1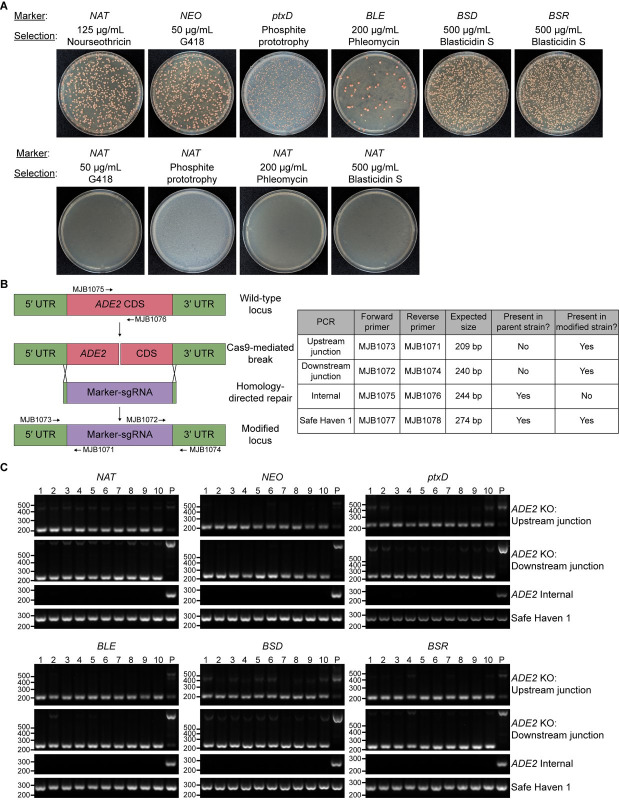 Fig 2
Fig 2| Marker | Selection | Experiment number | Transformants | Percentage of transformants relative
to |
|---|---|---|---|---|
|
| Nourseothricin (125 μg/mL) | 1 | 840 | 100.0 |
| 2 | 1,180 | 100.0 | ||
| 3 | 780 | 100.0 | ||
|
| G418 (50 μg/mL) | 1 | 670 | 79.8 |
| 2 | 1,310 | 111.0 | ||
| 3 | 1,300 | 166.7 | ||
|
| Phosphite prototrophy | 1 | 1,560 | 185.7 |
| 2 | 1,050 | 89.0 | ||
| 3 | 810 | 103.8 | ||
|
| Phleomycin (200 μg/mL) | 1 | 90 | 10.7 |
| 2 | 51 | 4.4 | ||
| 3 | 59 | 7.5 | ||
|
| Blasticidin S (500 μg/mL) | 1 | 3,270 | 389.3 |
| 2 | 4,000 | 339.0 | ||
| 3 | 2,530 | 324.4 | ||
|
| Blasticidin S (500 μg/mL) | 1 | 3,290 | 391.7 |
| 2 | 4,100 | 347.5 | ||
| 3 | 1,860 | 238.5 |
- —National Institute of Allergy and Infectious Diseaseshttp://dx.doi.org/10.13039/100000060
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFungal Infections and Studies · Antifungal resistance and susceptibility · RNA and protein synthesis mechanisms
OBSERVATION
Cryptococcus neoformans is an environmental fungus of both basic and biomedical significance (1). As a basidiomycete yeast that diverged from ascomycetes at least 450 million years ago (2), this organism encodes otherwise-conserved eukaryotic pathways that have been lost from traditional model fungi. Facile growth conditions and robust molecular genetics have made it an important model for dissecting such pathways (3–8). Clinically, C. neoformans causes opportunistic meningitis in immunocompromised individuals—particularly those with CD4^+^ T-cell deficiencies—leading to >118,000 annual deaths that include 19% of global HIV/AIDS-related mortality (9, 10). Despite its position in the “critical” tier of World Health Organization priority fungal pathogens (11), our understanding of how cryptococcal genes influence its infection biology remains in its infancy.
The haploid genome and defined sexual cycle of C. neoformans have made it well-suited for genetic studies. The development of biolistic transformation methods in the 1990s enabled the functional analysis of cryptococcal genes (12), including critical virulence factors. Construction of arrayed gene deletion libraries has driven systematic studies that have begun to build large-scale functional maps of this organism’s genome (8, 13–18). More recently, application of CRISPR/Cas9 tools to this system has streamlined genetic manipulation, replacing cumbersome biolistic protocols with electroporation and reducing the amount of homology necessary for gene replacement to just 50 bp (19–22). Further advances in robust genome-wide transposon mutagenesis (23), improved efficiency and accuracy of genetic modification (24, 25), and conditional knockdown systems (26, 27) are rapidly expanding the scope of genetic questions that can be probed.
Contemporary genetic manipulation of C. neoformans uses dominant markers to select for transformants. Drug resistance markers encoding hygromycin B phosphotransferase (HYG), neomycin phosphotransferase II (NEO), and nourseothricin acetyltransferase (NAT) confer resistance to hygromycin B, G418, and nourseothricin, respectively (28–30), and are the predominant selection methods used in the field. More recently, dominant prototrophic markers have been developed, with a marker encoding the Aspergillus nidulans acetamidase (amdS) enabling the use of acetamide as a sole nitrogen source (31), and a marker encoding the Pseudomonas stutzeri phosphite dehydrogenase (ptxD) allowing the use of phosphite as a sole phosphorus source (32). The amdS marker is particularly valuable because it confers sensitivity to fluoroacetamide and can therefore be counter-selected, enabling marker recycling or repair of disrupted loci (25, 31).
As the number of genetic tools in C. neoformans expands, the availability of additional selectable markers will facilitate its study. For example, the expression of Cas9 for CRISPR-based genome editing (22), TetR fusions for RNA-level gene regulation (26), or OsTir1 for auxin-dependent protein depletion (27) each requires the use of a marker. Combinations of such tools, along with knockouts and complementation of genes of interest, can quickly consume all available selection methods. While recycling amdS is one possible solution for multiple manipulations, this may not be optimal in all situations, such as when it has been used to temporarily delete genes that one may want to subsequently restore (25). Expanding the repertoire of dominant markers will thus enhance the complexity of genetic studies that can be performed in this organism.
Toward this end, we tested whether the commonly used selection drugs puromycin, phleomycin, and blasticidin S inhibit fungal growth on solid agar. We note that phleomycin was originally established for the selection of C. neoformans transformants 25 years ago (29) but has not, to our knowledge, been subsequently reported in the literature. While puromycin failed to inhibit fungal growth at concentrations up to 500 µg/mL, phleomycin and blasticidin S completely inhibited growth at 200 and 500 µg/mL, respectively (Fig. S1).
In a recent study, we found that electroporation of fused marker-guide PCR products containing overhangs for homology-directed repair (HDR) yielded near-perfect genome-editing efficiency when modifying a parent strain that both expresses Cas9 and lacks the non-homologous end joining (NHEJ) factor Ku80 (25). To determine whether phleomycin and blasticidin S could select for C. neoformans transformants, we expanded the marker-guide vector series reported in that work to include markers conferring resistance to phleomycin (bleomycin resistance gene [BLE]) and blasticidin S (blasticidin S deaminase [BSD] and blasticidin S resistance [BSR]) (Fig. 1). We also constructed a vector expressing the newly developed marker ptxD (32). In each case, we codon-optimized markers for C. neoformans expression and inserted an intron into an arbitrary location approximately one-third of the way through each coding sequence to improve transgene expression (22). As has been done with the most commonly used markers, we drove expression using the ACT1 promoter and the TRP1 terminator.
Vector series to produce marker-guide fusions using the BLE, BSD, BSR, or ptxD markers.
For each marker, we electroporated a Cas9-expressing, yku80-blaster parent strain with a PCR product containing (i) the marker of interest; (ii) an sgRNA targeting the coding sequence of the ADE2 gene; and (iii) 50 bp homology arms flanking the ADE2 coding sequence. All markers yielded transformants when plated onto their corresponding selection plates (Fig. 2A, top, and Table 1). “Mismatched” selection of NAT transformants on G418, phosphite, phleomycin, or blasticidin S plates failed to yield colonies (Fig. 2A, bottom), indicating an absence of spurious background colonies. Nearly all colonies developed red pigmentation characteristic of ADE2 deletion (Fig. 2A and Fig. S2), indicating efficient gene disruption. We confirmed this by isolating genomic DNA from 10 clones transformed with each marker and using PCR to assess (i) disruption of the ADE2 coding sequence and (ii) HDR at each end of the ADE2 coding sequence (Fig. 2B). Consistent with our previous work (25), all tested clones (10/10 colonies for all markers) lacked detectable ADE2 coding sequences and were positive for 5′ and 3′ junction PCR products characteristic of HDR (Fig. 2C).
Efficient disruption of ADE2 using novel and established selectable markers. A Cas9-expressing, yku80-blaster strain (CM2465) was electroporated with fused marker-guide constructs as described in the text. (A) Electroporated cultures were plated onto selection media corresponding to the appropriate marker (top row) or onto mismatched selection media to assess the frequency of background colonies (bottom row). Plates were incubated at 30°C for 3–4 days followed by storage at 4°C for 3 weeks to allow the red color characteristic of ade2Δ cells to emerge. (B) Diagram (not to scale) of the expected ADE2 modification by HDR (left) and table of expected diagnostic PCR results (right). (C) Diagnostic PCR assessing transformants for the presence of modified upstream and downstream ADE2 junctions and the absence of ADE2 coding sequence. The safe haven 1 locus serves as a positive control for PCR. Numbers above gels indicate transformant clone numbers. P indicates the untransformed parental strain.
TABLE 1: Transformants obtained in a Cas9-expressing, yku80-blaster background
We observed that the BLE marker reproducibly yielded fewer transformants compared to other markers (Table 1). We hypothesized that this might be due to increased sensitivity of our NHEJ-deficient parent strain to phleomycin, which induces double-stranded DNA breaks. However, the use of a Cas9-expressing parent strain with intact YKU80 did not improve transformant yield to the level of a NAT control (Table S1). Reducing the phleomycin selection concentration from 200 to 100 μg/mL yielded only a modest (~2-fold) increase in the number of transformants obtained (Table S2). These data suggest that, while BLE can effectively select for transformants, it does so with lower efficiency than other markers under these conditions.
In summary, we have expanded the number of dominant selection methods available for C. neoformans to seven, including three well-established selection drugs (hygromycin B, G418, and nourseothricin), two dominant prototrophic methods (acetamide and phosphite prototrophy), one newly established selection drug (blasticidin S), and one previously reported yet unutilized selection drug (phleomycin). We have adapted these to be compatible with marker-guide fusion DNA constructs that we have recently demonstrated to enhance homology-dependent genome-editing efficiency, especially when combined with a reversible Ku80 mutation. With the rapidly expanding genetic toolbox for C. neoformans, the availability of multiple robust and validated selection methods will provide significant flexibility in constructing strains incorporating multiple sequential modifications.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1May RC, Stone NRH, Wiesner DL, Bicanic T, Nielsen K. 2016. Cryptococcus: from environmental saprophyte to global pathogen. Nat Rev Microbiol 14:106–117. doi:10.1038/nrmicro.2015.626685750 PMC 5019959 · doi ↗ · pubmed ↗
- 2Berbee ML, Taylor JW. 2010. Dating the molecular clock in fungi – how close are we? Fungal Biol Rev 24:1–16. doi:10.1016/j.fbr.2010.03.001 · doi ↗
- 3Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, Moresco JJ, Yates JR III, Bartel DP, Madhani HD. 2013. Stalled spliceosomes are a signal for RN Ai-mediated genome defense. Cell 152:957–968. doi:10.1016/j.cell.2013.01.04623415457 PMC 3645481 · doi ↗ · pubmed ↗
- 4Dumesic PA, Homer CM, Moresco JJ, Pack LR, Shanle EK, Coyle SM, Strahl BD, Fujimori DG, Yates JR III, Madhani HD. 2015. Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 160:204–218. doi:10.1016/j.cell.2014.11.03925533783 PMC 4303595 · doi ↗ · pubmed ↗
- 5Burke JE, Longhurst AD, Merkurjev D, Sales-Lee J, Rao B, Moresco JJ, Yates JR III, Li JJ, Madhani HD. 2018. Spliceosome profiling visualizes operations of a dynamic RNP at nucleotide resolution. Cell 173:1014–1030. doi:10.1016/j.cell.2018.03.02029727661 PMC 5940017 · doi ↗ · pubmed ↗
- 6Catania S, Dumesic PA, Pimentel H, Nasif A, Stoddard CI, Burke JE, Diedrich JK, Cooke S, Shea T, Gienger E, Lintner R, Yates JR III, et al.. 2020. Evolutionary persistence of DNA methylation for millions of years after ancient loss of a de novo methyltransferase. Cell 180:263–277. doi:10.1016/j.cell.2019.12.01231955845 PMC 7197499 · doi ↗ · pubmed ↗
- 7Sales-Lee J, Perry DS, Bowser BA, Diedrich JK, Rao B, Beusch I, Yates JR III, Roy SW, Madhani HD. 2021. Coupling of spliceosome complexity to intron diversity. Curr Biol 31:4898–4910. doi:10.1016/j.cub.2021.09.00434555349 PMC 8967684 · doi ↗ · pubmed ↗
- 8Boucher MJ, Banerjee S, Joshi MB, Wei AL, Nalley MJ, Huang MY, Lei S, Ciranni M, Condon A, Langen A, Goddard TD, Caradonna I, et al.. 2025. Phenotypic landscape of an invasive fungal pathogen reveals its unique biology. Cell 188:4003–4024. doi:10.1016/j.cell.2025.05.01740505656 PMC 12407185 · doi ↗ · pubmed ↗