The Development of Stereoselective Substrate and Reagent-Controlled Lithiation–Borylation Chemistry
Yannick Linne, Maike Birkner, Daniel Lücke, Jan Flormann, Kjeld Gerdes, Giada Tedesco, Gaia Stojanovic, Tom Jentsch, Birk Jäger, Kevin Bajerke, Jörg August Becker, Markus Kalesse

TL;DR
This paper introduces a new method for creating allylic alcohols in a stereoselective way using chiral polyketide fragments.
Contribution
A novel stereoselective lithiation–borylation protocol using chiral polyketide fragments without sparteine.
Findings
Chiral polyketide fragments enable high-yield and diastereoselective Hoppe–Matteson–Aggarwal rearrangements.
Steric and electronic effects strongly influence stereochemical outcomes.
Guidance flowcharts were developed using conformational analysis and a Felkin-like model.
Abstract
Allylic alcohols are a privileged motif in polyketide-based natural product synthesis, and new methods that access them in a stereoselective fashion are highly sought after. Toward this goal, we found that the use of chiral polyketide fragments allows for performing the Hoppe–Matteson–Aggarwal rearrangement in the absence of sparteine with high yields and diastereoselectivities, rendering this protocol a highly valuable alternative to existing methods. Various stereodyads and -triads bearing different protecting and directing groups were investigated to determine their substrate induction. The mostly strong inherent induction was attributed to either steric or a combination of steric and electronic effects. The stereochemical outcome could be explained by (DFT-based) conformational analysis and a Felkin-like model, allowing guidance flowcharts to be created for the substrate- and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1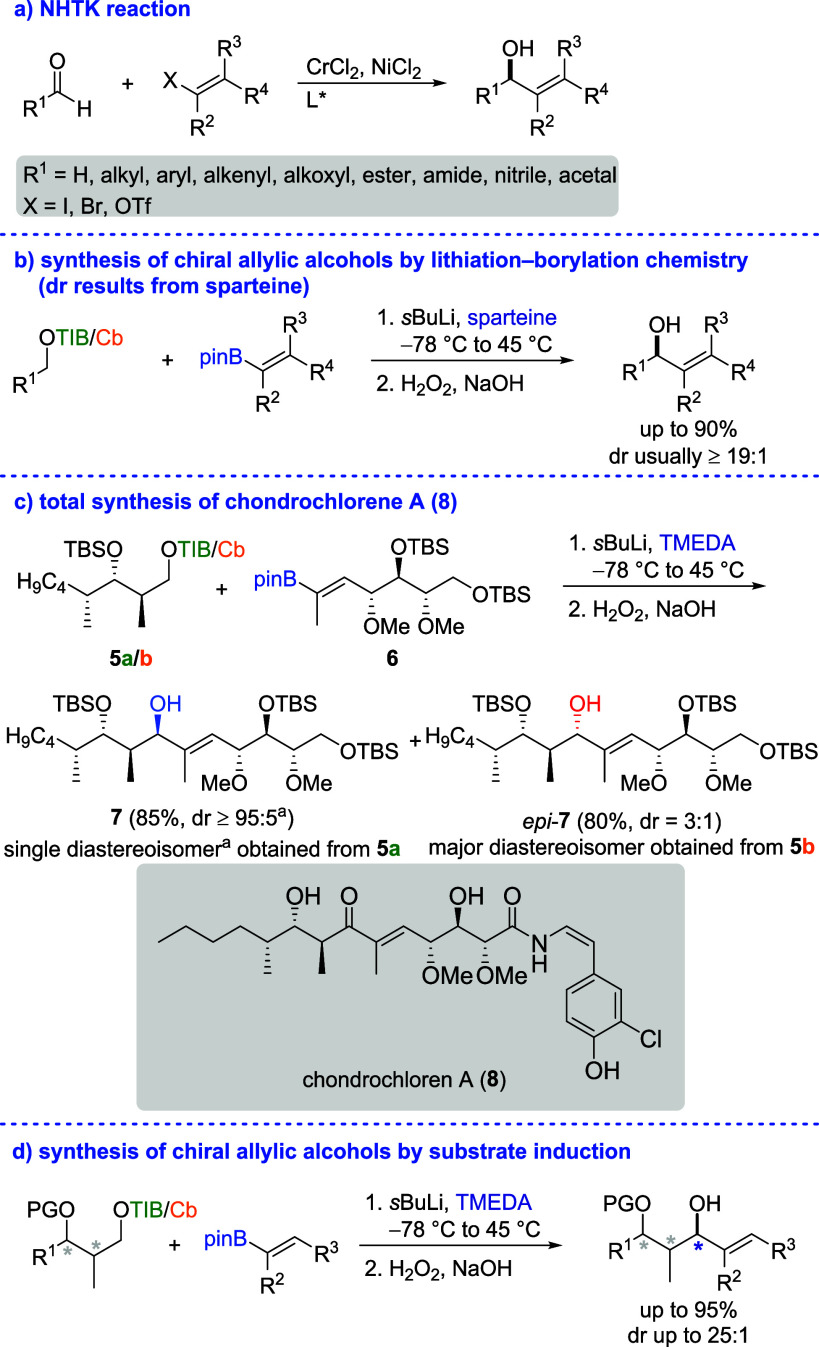 1
1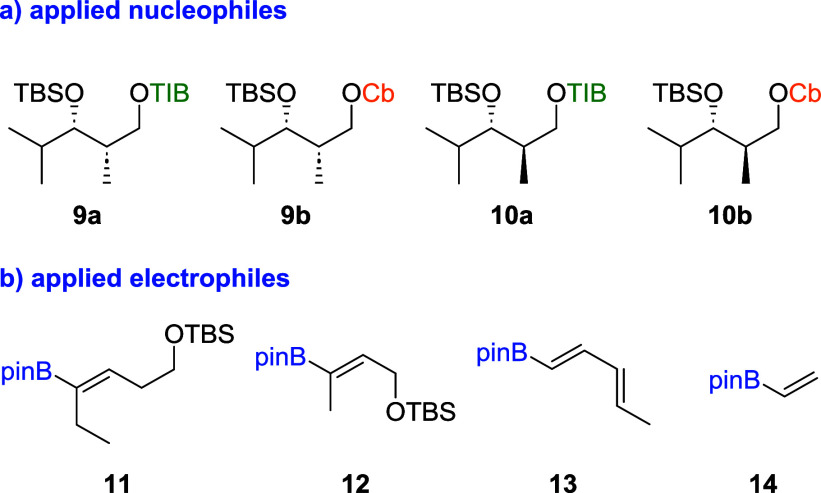 2
2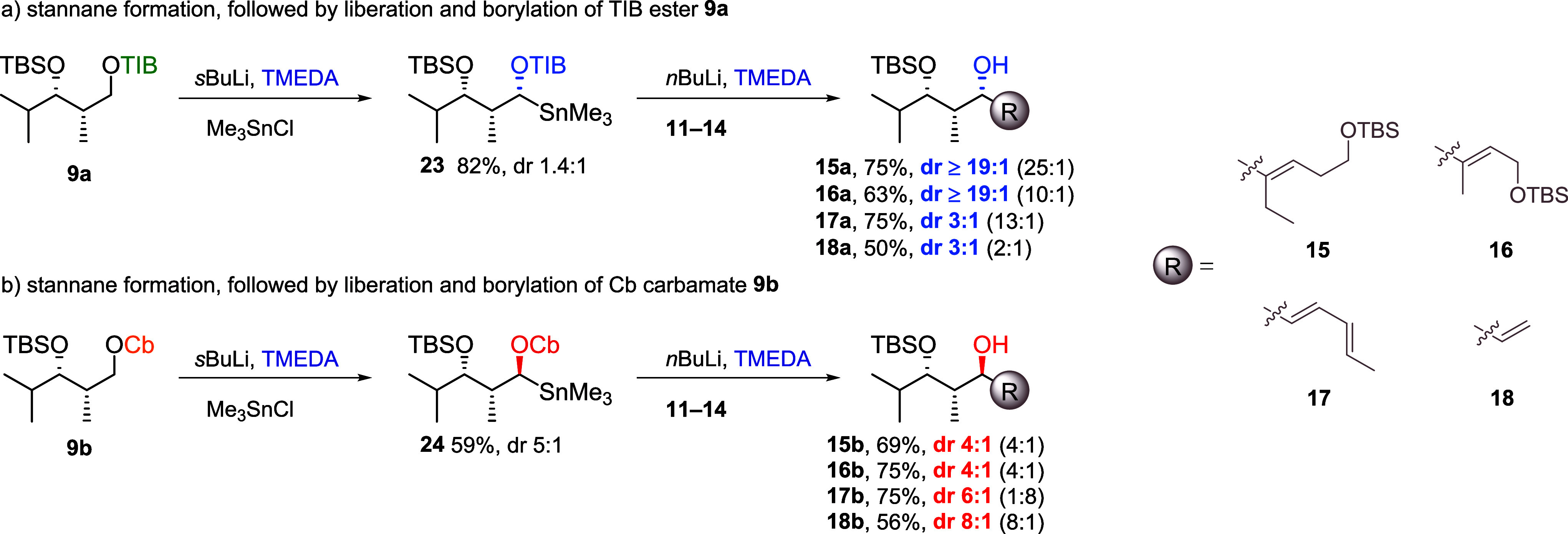 2
2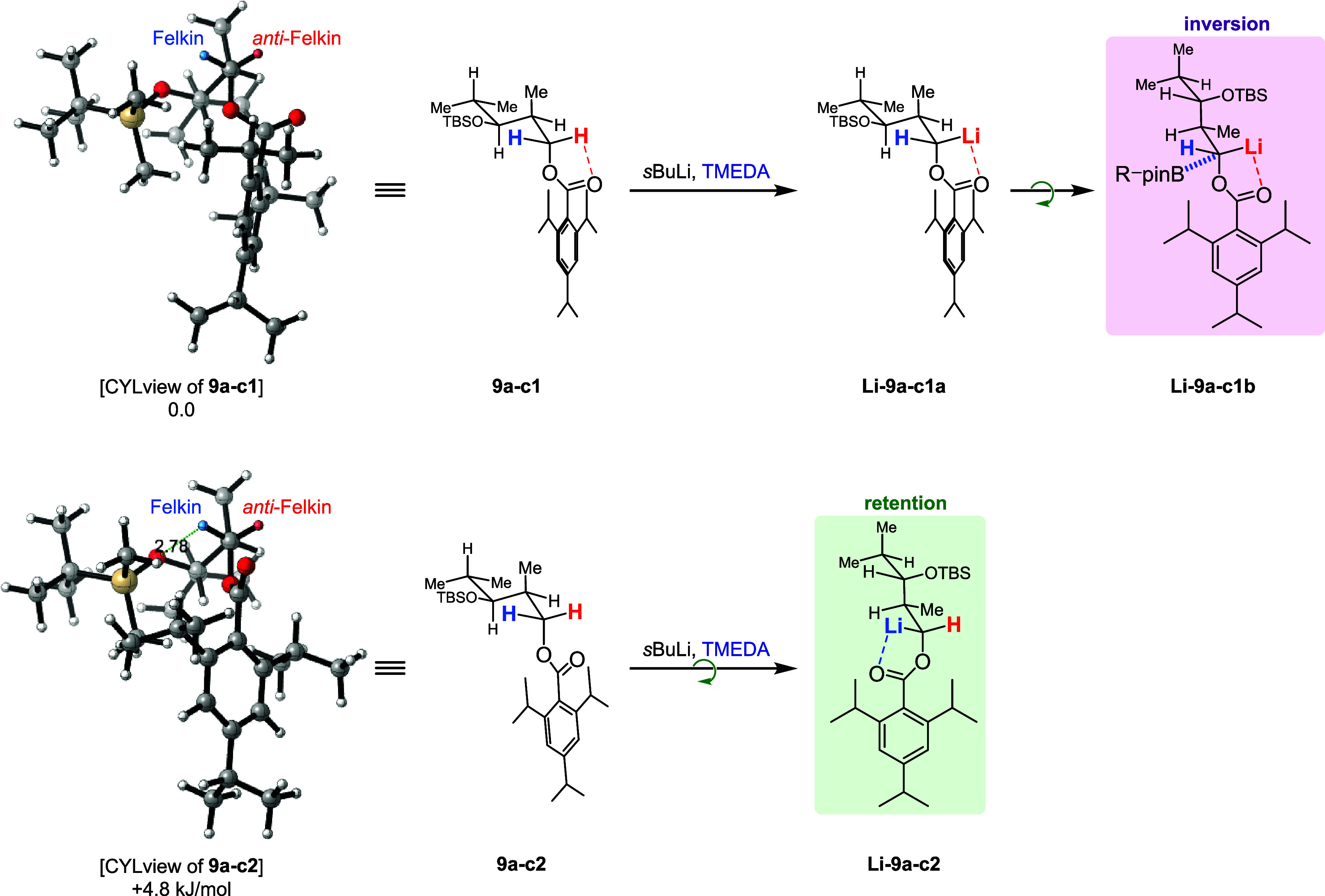 3
3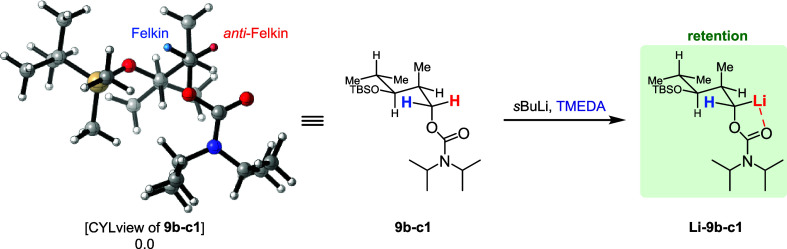 4
4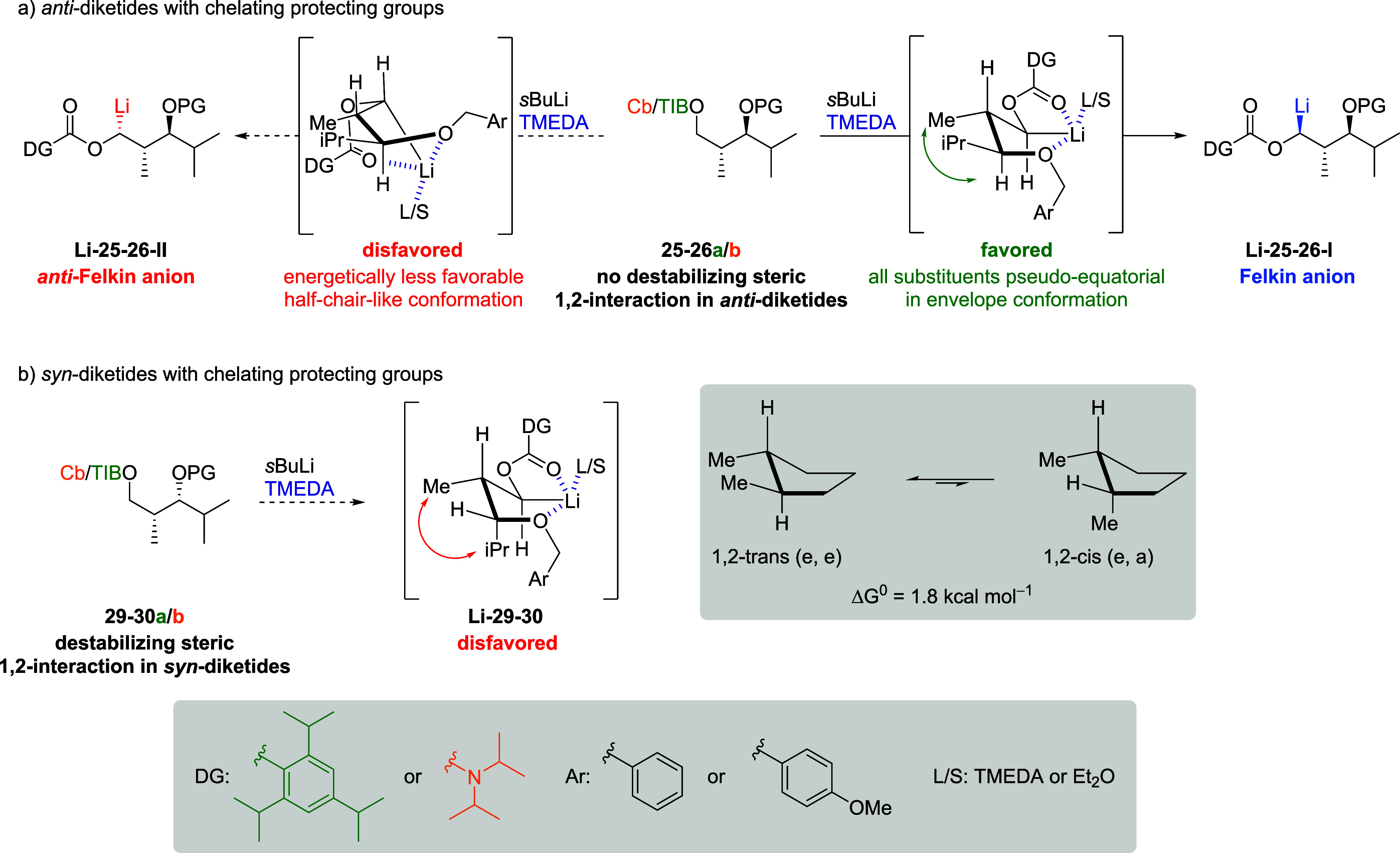 5
5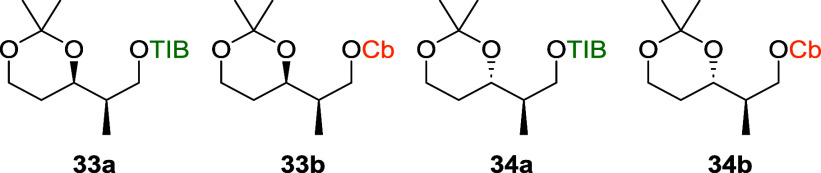 3
3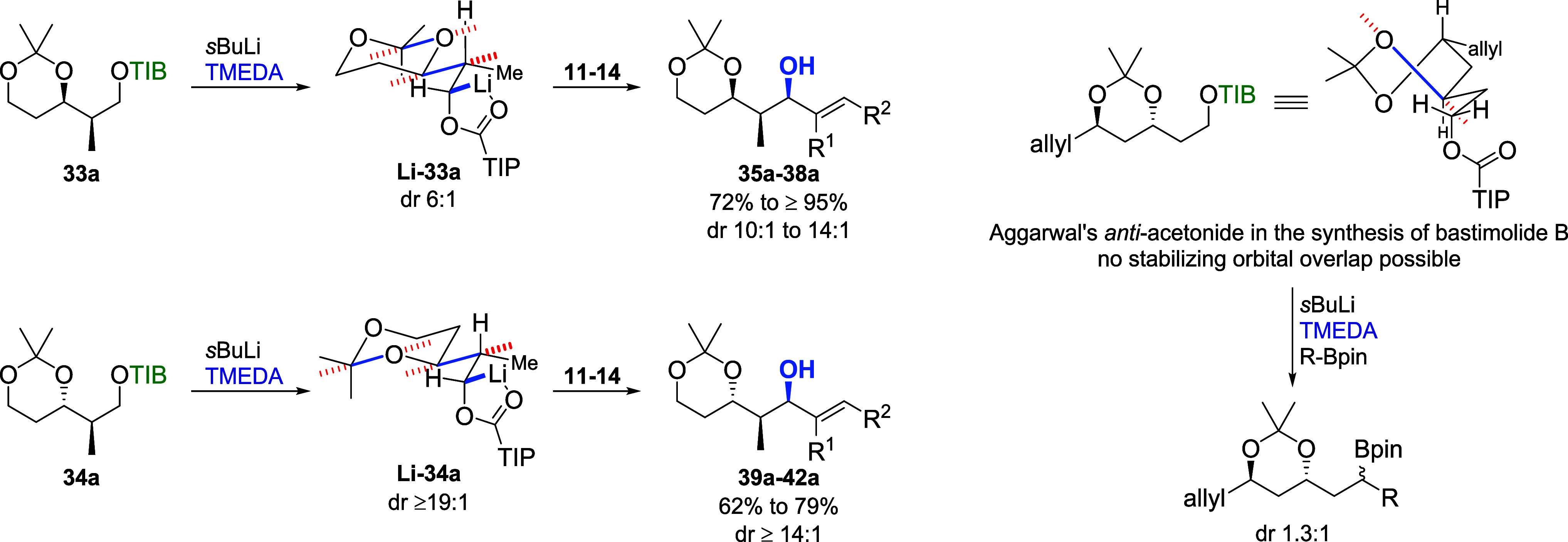 6
6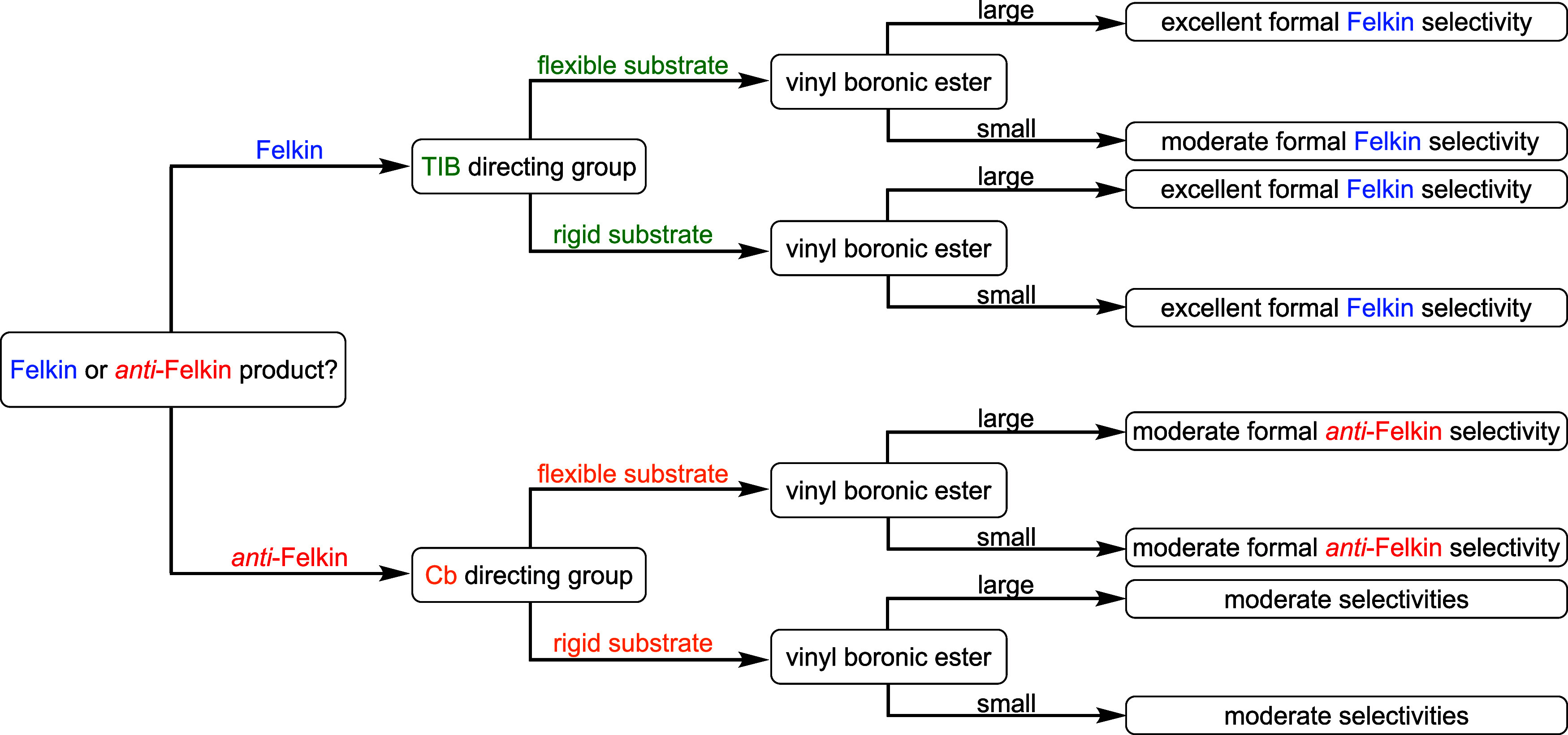 4
4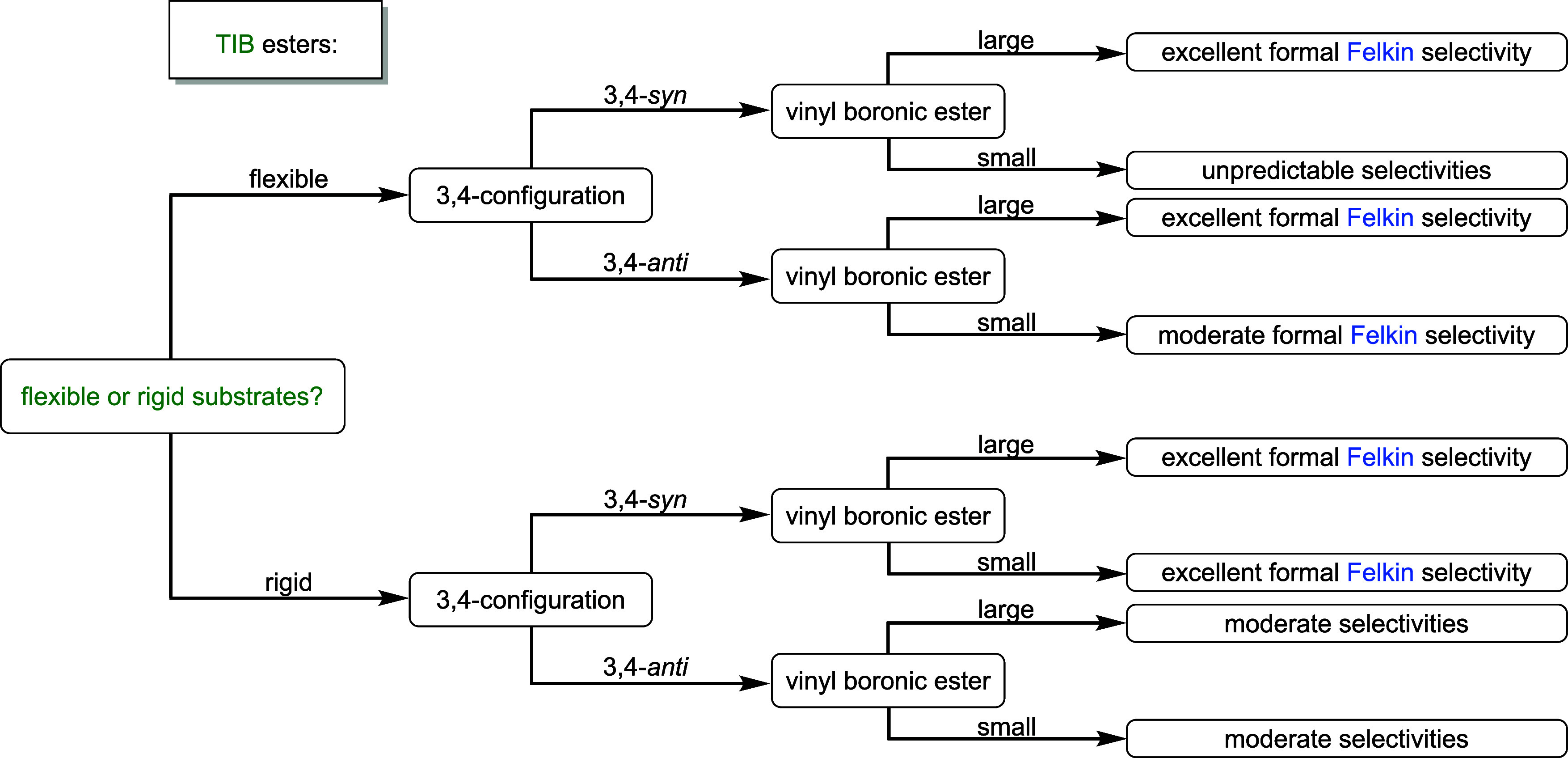 5
5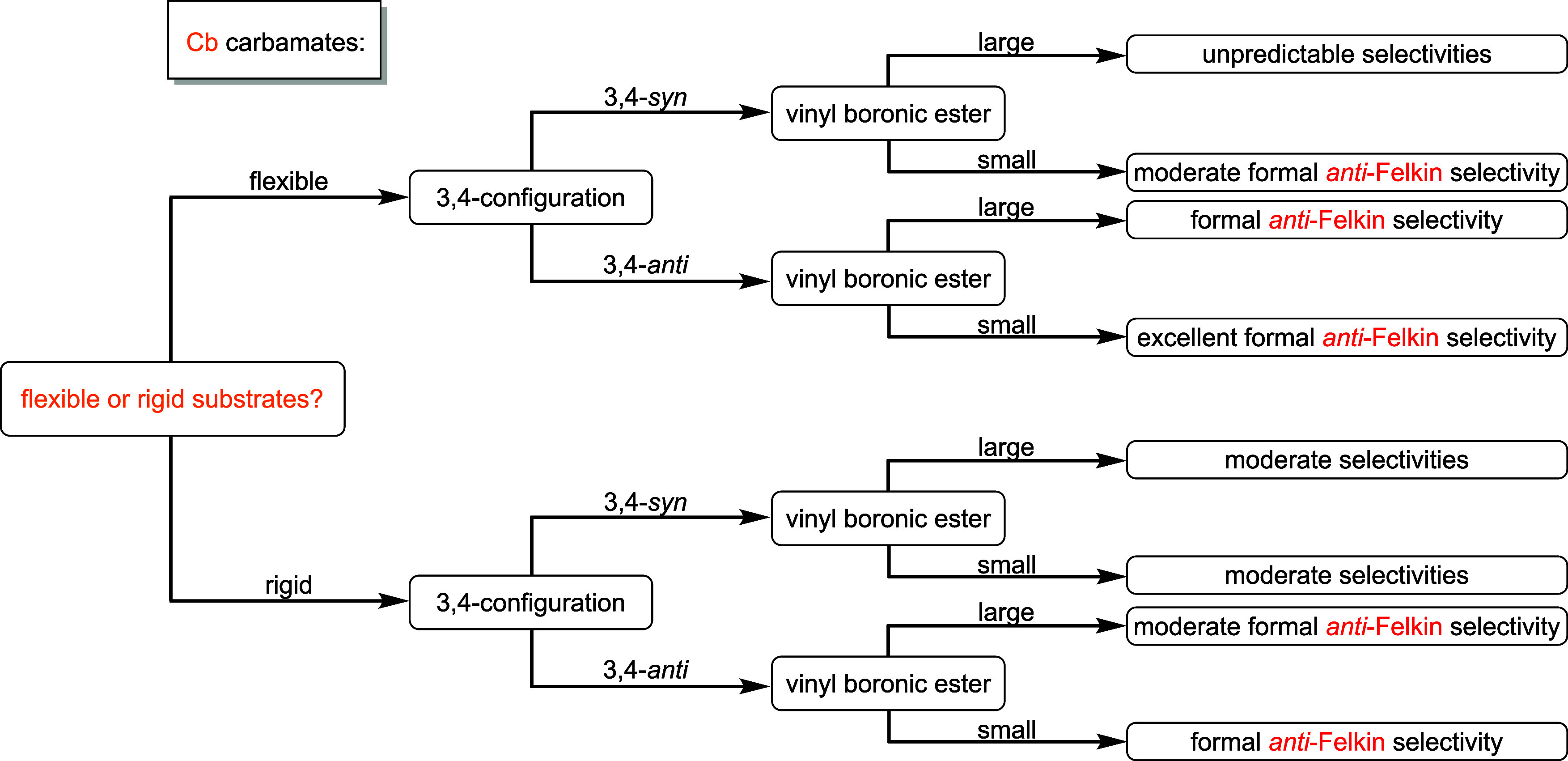 6
6- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Synthetic Organic Chemistry · Asymmetric Synthesis and Catalysis · Synthetic Organic Chemistry Methods
Introduction
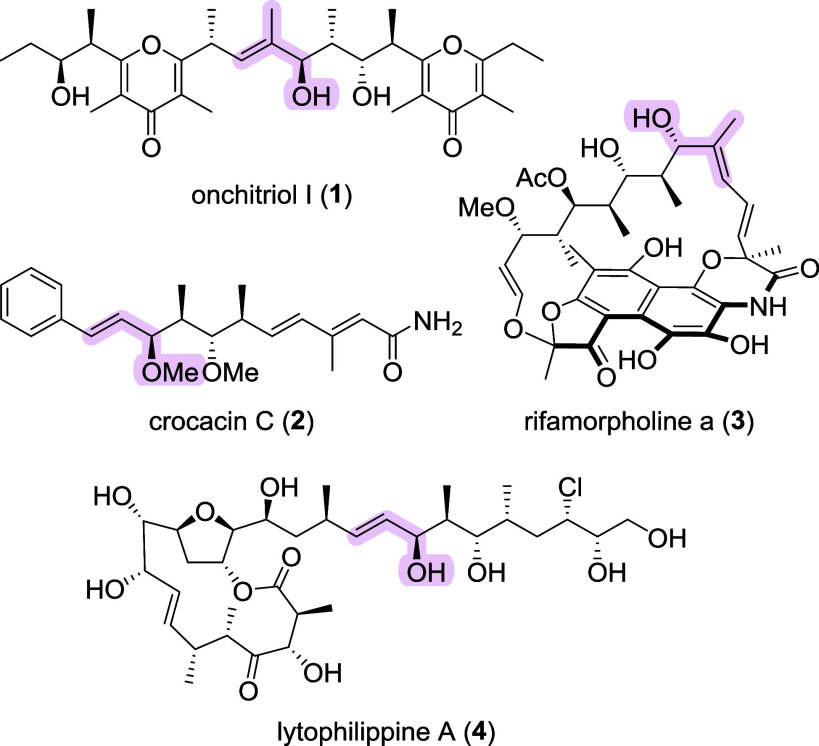
Chiral allylic alcohols are prominent structural motifs in natural products (Figure)? as well as versatile synthetic building blocks.
Natural products bearing chiral allylic alcohols or their derivative motifs.
A general way of accessing these motifs is the addition of a vinyl organometallic species to an aldehyde. Among other protocols, the Nozaki–Hiyama–Takai–Kishi reaction (NHTK reaction, Schemea)? is the method of choice for the asymmetric synthesis of chiral allylic alcohols. The high chemoselectivity toward aldehydes tolerates various other functional groups like ketones, acetals, nitriles, or esters. However, the obtained yields and selectivities are often moderate, especially for more complex substrates. ?,?,?,? We recognized that the chemistry of lithiation and borylation developed by Hoppe, Matteson, and Aggarwal? could serve as a platform for the stereoselective synthesis of chiral allylic alcohols. In contrast to the NHTK reaction, the polarities are interchanged as a vinyl boronic ester acts as the electrophile and is attacked by the anion of a masked alcohol (Hoppe anion). Even though the Hoppe–Matteson–Aggarwal rearrangement has been applied to the synthesis of numerous natural products,? no example for the synthesis of an allylic alcohol was given in the literature before we enrolled our program. During our research, we developed a protocol for the synthesis of chiral allylic alcohols by lithiation–borylation chemistry (Schemeb)? and applied it in our total synthesis of the polyketide-peptide hydrid chondrochloren A (8) (Schemec).? Interestingly, the linkage of vinyl boronic ester (VBE) 6 and 2,4,6-triisopropylbenzoyl (TIB) ester 5a provided alcohol 7 as a single diastereoisomer in the presence of achiral *N,N,N′,N′-*tetramethylethylenediamine (TMEDA), indicating a strong substrate induction for this transformation. Surprisingly, the formation of the opposite diastereoisomer epi-7 was favored, when the TIB group was replaced by a N,N-diisopropyl carbamoyl (Cb) group. Even though the stereochemistry of the obtained secondary alcohol was inconsequential for our synthesis as it was oxidized later on, this finding served as a starting point for further investigations on directing effects in 1,2-metalate rearrangements (Schemed).? Apart from our striving for a deeper understanding of the origin of asymmetric induction, we also noticed the potential for even more synthetic applications of lithiation–borylation chemistry, if high levels of diastereoselectivity could be obtained in the absence of the expensive chiral diamine sparteine.?
Synthesis of Chiral Allylic Alcohols by the NHTK Reaction and Lithiation–Borylation Chemistry
Herein, we report a detailed overview of our investigations on substrate induction in lithiation–borylation chemistry for the synthesis of allylic alcohols. Parts of this work were previously published? and are included to provide an overall overview. Furthermore, flowcharts for synthetic applications are given.
Results and Discussion
We started our investigations with isobutyraldehyde derived diketides 9a/b and 10a/b as their isobutyl residue resembles a polyketidal framework (Figure). In our synthesis of chondrochloren A (8), we obtained a switch in selectivity when altering the directing group from TIB to Cb.? To examine whether this is a general trend, both directing groups were utilized throughout the entire study. In addition, the influence of different conformations originating from a syn as well as an anti relation between the protected alcohol and the methyl branch was investigated. Vinyl boronic esters 11–14 were used as electrophiles (Figure). The more complex VBEs 11–13 representing the situation of branched/complex double bonds present in many natural products, whereas vinylboronic acid pinacol ester (14) was chosen to determine whether the size of the electrophile has an impact on the diastereoselectivity of the reaction.
Nucleophiles and electrophiles applied to the first study.
For all reactions with the anion derived from TIB diketide 9a, the formal Felkin products were obtained as the major diastereoisomers (Table, entries 1, 3, 5, and 7). Very good yields were obtained for branched vinyl boronic esters 11 and 12 (86 and 79%), whereas 13 and 14 provided the corresponding allylic alcohols in moderate yields (45 and 50%). In terms of selectivity, better results were obtained with the more complex VBEs 11–13 (dr starting from 10:1) compared to vinylboronic acid pinacol ester (14) (entry 7, dr 2:1). The low selectivity and moderate yield of the reaction of 9a and 14 could be increased by the use of (+)-sparteine (entry 8), indicating a matched situation for this transformation. This was further supported when (+)-sparteine was replaced by its enantiomer (−)-sparteine (entry 9). In theory, the exchange of enantiomers should lead to the formal anti-Felkin product; however, formal Felkin product 18a was obtained as the major diastereoisomer. The decrease in selectivity (dr 3:1) and yield (16%) indicates a mismatched situation. As both enantiomers of sparteine favor the formation of diastereomeric anions,? this observation serves as a first indicator for an inversion during the borylation step. In comparison to TIB ester 9a, lithiation–borylation of Cb carbamate 9b proceeded in lower yields (entries 2, 4, 6, and 10). Branched VBEs 11 and 12 led to the formal anti-Felkin products in moderate yields (41 and 38%) and selectivities (4:1). Surprisingly, formal Felkin product 17a was obtained in a good selectivity (dr 8:1) but very low yield (3%) from the reaction of dienyl boronic ester 13. Transformation with less complex VBE 14 (entry 10) provided the formal anti-Felkin product in a good selectivity (dr 8:1) and moderate yield (31%). The anti-Felkin selectivity could be increased by the use of (−)-sparteine (entry 12). Interestingly, the use of (+)-sparteine resulted in the expected formal Felkin product 18a as the sole diastereoisomer (entry 11), indicating a preference for retention during the borylation of Cb carbamates.?
1: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of syn-Diketides 9a/b
Comparable results were obtained when anti-diketides 10a/b were applied to lithiation–borylation conditions (Table). For TIB ester 10a, the formal Felkin products were obtained in all cases (entries 1, 3, 5, and 7). Again, branched vinyl boronic esters 11 and 12 performed well providing excellent selectivities (≥19:1) along with good to very good yields (79 and 69%). The reactions of linear VBEs 13 and 14 provided good to moderate yields (69 and 50%) and lower selectivities (dr 5:1 and 2:1). With Cb carbamate 10b, the formal anti-Felkin product was obtained in all cases. Compared to the formal Felkin products, lower yields (11–60%) and much lower selectivities (approximately 2:1 for all VBEs) were obtained. It was again possible to fully override the anti-Felkin selectivity of the Cb carbamate for the reaction with VBE 14 when (−)-sparteine was used instead of TMEDA (entry 12, dr 19:1). Interestingly, all other reactions of anti-diketides 10a/b with 14 in the presence of either enantiomer of sparteine (entries 8, 9, and 11) did not lead to any product formation, indicating a strong dependency between conformation of the (Hoppe) anion and the size of the diamine ligand.?
2: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of anti-Diketides 10a/b
For a better understanding whether the stereoselectivity originates from the lithiation or borylation step, the carbanions of all diketides were trapped as their corresponding trimethylstannanes.? This transmetalation process is known to proceed under full retention,? which is a general trend for the reaction of nonmesomerically stabilized Hoppe anions with electrophilic reagents (e.g., TMSCl, CO_2_, Me_3_SnCl, MeI, or boronic acids). ?−? ? ? Thus, the diastereomeric ratios of the obtained stannanes should reflect the diastereomeric ratios of the carbanions. TIB ester 9a-derived stannane 23 (Schemea) was obtained in a low diastereoselectivity of 1.4:1 favoring the fomal Felkin product (configurations of all stannanes were determined by deprotonation with (+)-sparteine and (−)-sparteine, followed by addition of Me_3_SnCl; for more details, see the Supporting Information). The obtained low selectivity does not parallel the excellent results obtained for the reaction of the same carbanion with VBEs 11–13 (Table, entries 1, 3, and 5). This indicates that the overall stereoselectivity of this transformation is not controlled during lithiation, but the borylation step. For the selective formation of the formal Felkin product, the ate-complex formation most likely takes place under inversion and retention depending on the configuration of the carbanion. To further support this hypothesis, stannane 23 was subjected to Sn–Li exchange followed by addition of VBEs 11–14. In all cases, the diastereomeric ratio of the obtained allylic alcohols increased compared to the applied stannanes, supporting the hypothesized inversion processes during the ate-complex formation step. The obtained selectivities of allylic alcohols 15a, 16a, and 18a paralleled the results of the one-pot reaction. However, the formation of 17a proceeded in a lower diastereoselectivity. These observations are in line with previous reports by Hoppe and Schleyer describing inversion and retention processes during the reaction of benzylic carbanions.? They mentioned that electronic as well as steric effects determine which mechanistic pathway is chosen. The strong influence of sterics is highlighted in our work by the excellent selectivities obtained with sterically demanding electrophiles 11 and 12. Stannane 24 derived from Cb carbamate 9b was obtained in a diastereomeric ratio of 5:1 favoring the formal anti-Felkin product (Schemeb), which parallels the previously observed selectivities. Reliberation of the carbanion followed by borylation with VBEs 11–14 gave access to allylic alcohols 15b–18b. The obtained diastereoselectivities of 15b–17b are in accordance with 24, indicating that the ate-complex formation takes place under retention of configuration. The higher formal anti-Felkin selectivity of 18b can either be explained by partial inversion during the ate-complex formation or by the preferred reaction of the formal anti-Felkin carbanion. A preferred reactivity of this carbanion and partial decomposition of nonreacting formal Felkin carbanion could also serve as an explanation for the lower yield obtained for this transformation. Similar results were obtained for the carbanions derived from diketides 10a/b (see the Supporting Information for more details), providing additional support for inversion and retention processes during the ate-complex formation of TIB ester-derived anions and mainly retention for the reaction of their Cb counterparts. A stronger tendency toward retention during the ate-complex formation of carbamate-derived anions could also be explained by a hypothesis made by Beak, who reported that ″highly reactive or nonlithium coordinating electrophiles proceed with inversion, while less reactive and lithium coordinating electrophiles give retention″.? In our case, electrophile is exchanged by nucleophile, so both of Beak’s criteria could serve as an explanation for the observed reactivities. The lower reactivity of the Cb carbamates compared to the TIB esters is reflected by the need of MgBr_2_·OEt_2_ as an additional Lewis acid and elevated temperatures to initiate the 1,2-metalate rearrangement. The stronger coordination of lithium can be rationalized with the amide resonance.
Stannane Formation Followed by Liberation and Borylation of 9a/b
To be able to discuss the differences between the diastereomeric ratio of the formed carbanions and the obtained allylic alcohols in more detail, we decided to initiate a quantum chemical search for all four diketides (9a/b and 10a/b). The results of this computational analysis were discussed in our initial study? and highlighted 9a–c1 and 9a–c2 as the most stable conformers of TIB ester 9a at −78 °C. For both conformers, the carbonyl oxygen of the TIB ester points toward the formal anti-Felkin proton. This is in line with the assumption that the base is coordinated and directed by the carbonyl group prior to lithiation. In 9a–c1, the proton in the anti-Felkin hemisphere is more easily accessible, which would lead to a preferred formation of anion Li-9a-c1a. However, in Li-9a-c1a, both hemispheres are difficult to access by an electrophile as the anti-Felkin hemisphere is shielded by the 2,4,6-triisopropylphenyl (TIP) group and the Felkin hemisphere is blocked by the isobutanol residue. A slight conformational change to Li-9a-c1b could lead to a free Felkin hemisphere supporting an inversion during the ate-complex formation. In conformer 9a-c2, which is higher in energy than 9a-c1 but also populated at −78 °C, it is most likely that the conformation changes before deprotonation of the formal Felkin proton as the Felkin hemisphere is otherwise too hindered. Formed anion Li-9a-c2 could then perform the borylation under retention of its configuration. The preferred deprotonation of the formal Felkin proton in 9a-c2 might be explained by its close proximity to the oxygen of the TBS-ether. An increased basicity/reactivity of protons close to silyl ethers was previously described by Knochel et al.?
In the case of Cb carbamate 9b, the most stable conformer at −78 °C is 9b-c1. Here, the *anti-*Felkin hemisphere is easier to access leading to Li-9b-c1 as the preferentially formed anion, which could then react under retention of its configuration.
As the obtained yields and selectivities for the reactions of vinylboronic acid pinacol ester (14) were below all other VBEs, we decided to further optimize this transformation. During their synthesis of stemaphylline, Aggarwal and co-workers showed that a solvent swap drastically increased the yield of a 1,2-metalate rearrangement,? so we decided to perform a solvent screening on the aforementioned transformation. Anti-diketides 10a/b were chosen for the optimization studies as they performed weaker than their syn-analogs (Table, entries 7–12 vs Table, entries 7–12). When TIB ester 10a was applied, a solvent switch from diethyl ether (previous conditions, Table, entry 1) to tetrahydrofuran (THF, entry 2) for the addition of 14 provided a comparable yield of 47% along with a much better diastereoselectivity of 6:1 favoring the formal Felkin product. Performing the entire reaction in THF, however, led to no product formation (entry 3). To keep the reaction operationally simple, we continued searching for a solvent performing well in the deprotonation as well as the metalate rearrangement step. Cyclopentyl methyl ether (CPME), which provided very good results in the study of Aggarwal et al.,? led in our case only to traces of the product (entry 4). An increase in yield to 60% along with a diastereoselectivity of 5:1 favoring the formal Felkin product was obtained when the reaction was performed in methyl tert-butyl ether (MTBE, entry 5). Using a nonpolar solvent for the deprotonation (toluene) followed by addition of 14 in THF did not provide any product (entry 6). For Cb carbamate 10b, only MTBE turned out to be suitable for the formation of formal anti-Felkin product 22b (entry 11). However, the obtained yield and selectivity was slightly lower than our previous result (entry 7). With all other solvent combinations, no product formation or only traces of 22b were observed (entries 8–10 and 12).
3: Solvent Screening for the Synthesis of Allylic Alcohols 22a/b
We were also interested if the protecting group of the neighboring alcohol would have a strong impact on the substrate induction. Previous work by Hoppe showed that excellent diastereoselectivies could be obtained for the alkylation of carbanions if a chelating five-membered acetonide was present.? Again, the reaction of VBE 14 with different protected anti-configured diketides was investigated first (Table). In comparison to our previous investigations (Table, entries 7–12), the tert-butyldimethylsilyl (TBS)-protecting group was replaced by a theoretically stronger chelating benzyl (Bn) or para-methoxybenzyl (PMB) group. For TIB diketides25a and 26a, the formal Felkin products 27a and 28a were obtained in excellent selectivities (entries 1 and 3, dr 19:1) along with moderate yields of 45 and 54%, respectively. When the corresponding Cb carbamates 25b and 26b were used, the obtained major diastereomers were the formal Felkin products, respectively (entries 2 and 4), supporting an intramolecular coordination of the carbanion. However, the excellent selectivity of 19:1 was only obtained in the case of the PMB ether. In line with our previous results for the reaction of Cb carbamates with VBE 14 (Table and ?, entries 10–12), low yields of 10% (Bn ether) respectively 4% (PMB ether) were obtained.
4: Screening of Different Protecting Groups for the Reaction of anti-Configured Diketides with VBE 14
Due to the improved selectivities obtained for the anti-configured diketides, their syn counterparts were investigated next (Table). TIB diketides 29a and 30a provided the formal Felkin products 31a and 32a in moderate yields along with moderate to low selectivities (entries 1 and 3). For Cb carbamates 29b and 30b, the formal anti-Felkin products 31b and 32b were obtained in low yields and moderate to good selectivities. These selectivities are comparable to the previously obtained results of TBS protected diketides 9a/b (Table, entries 7 and 10) indicating less influence through coordination.
5: Screening of Different Protecting Groups for the Reaction of syn-Configured Diketides with VBE 14
The difference in selectivity of the applied anti- and syn-diketides might be explained by intermediate carbanions (Scheme). Similar to the work of Hoppe,? we propose intramolecular lithium coordination by the carbonyl group and the ether oxygen for anti-diketides 25a/b and 26a/b leading to bicyclic Li-25–26-I as favored carbanion. In line with experimental results, this carbanion would lead to the formal Felkin products. Diasteromeric carbanion Li-25–26-II might be formed as well but should be disfavored since for abstraction of the anti-Felkin proton, the chelate would have to adopt an energetically less favorable half-chairlike conformation, which is in line with previous observations of cyclic five-membered carbanions described by Knochel et al.? For syn-diketides 29a/b and 30a/b double coordination would lead to carbanion Li-29–30. However, 1,2-repulsion between the isopropyl and methyl group? might favor carbanions similar to 9a/b (Schemes and ?), which would also explain the similarity of selectivities (Table, entries 7 and 10, and Scheme).
Rationalization of Inversion and Retention during the Borylation of TIB Ester 9a
Rationalization of Retention during the Borylation of Cb Carbamate 9b
Proposed Carbanions Rationalizing the Observed Selectivities of Diketides 25, 26, 29, and 30
Interested in the effect of a less flexible conformation, we decided to investigate the substrate induction of acetonide-protected diketides 33a/b and 34a/b with VBEs 11–14 next (Figure).?
Investigated acetonide-protected diketides.
In contrast to the abovementioned five-membered acetonide utilized by Hoppe,? we chose six-membered acetonides as their functional group distance parallels the situation in polyketides. Additionally, the acetonides in 33a/b and 34a/b do most likely adopt the chair conformation, so their level of substrate induction could be compared to previous work done by the Aggarwal group. In their synthesis of bastimolide B, the substrate induction of a six-membered anti-acetonide (twist-boat conformation) was investigated, which unfortunately showed nearly no asymmetric induction.? For syn-configured TIB ester 33a, the formal Felkin products 35a–38a (Table, entries 1, 3, 5, and 7) were obtained in good yields (starting from 72%) and very good selectivities (dr starting from 10:1). This time, the reaction with vinylboronic acid pinacol ester (14) provided similar results as with all of the other VBEs. Surprisingly, Cb carbamate 33b did as well provide the formal Felkin products although in lower selectivities (entries 2, 4, 6, and 8). In the presence of (+)-sparteine, the reaction of 33b with 14 (entry 9) provided the formal anti-Felkin product 38b in a moderate selectivity of 4:1 along with a low yield of 17%. In contrast to our previous results (Tables and ?), this was the first time that the selectivity for the reaction of a carbamate in the presence of sparteine did not provide a dr of 19:1, indicating a mismatched situation between substrate and reagent control. However, for the reaction with (−)-sparteine (entry 10), the previously obtained excellent selectivities of 19:1 could be restored.?
6: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of syn-Diketides 33a/b
Compared to its syn-configured counterpart, anti-configured TIB ester 34a provided the formal Felkin products 39a–42a (Table, entries 1, 3, 5, and 7) in even higher selectivities (dr starting from 14:1) but lower yields (starting from 62%). For the reactions of Cb carbamate 34b with more complex vinyl boronic esters 11–13 (entries 2, 4, and 6), the formal anti-Felkin products were obtained in moderate to good yields (52–66%) and low to moderate selectivities (dr 1:1.3–1:4). The reaction with 14 (entry 8), however, led to formal Felkin product 42a in a low yield of 10% along with a low selectivity of 2:1. The presence of (+)-sparteine (entry 9) could fully overrule the substrate induction as formal anti-Felkin product 42b was obtained in an excellent selectivity of 1:19 but a low yield of 7%. Interestingly, the presence of (−)-sparteine (entry 10) did not favor the formation of any diastereoisomer (dr 1:1).?
7: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of anti-Diketides 34a/b
This time, the origin of the diastereoselectivity was investigated by deuteration of the carbanions of 33a/b and 34a/b (see the Supporting Information).? For 33a, a dr of 6:1 was determined indicating that the borylation occurs again under retention and inversion. The diastereomeric ratio obtained for deuterated 33b was 4:1, which is in good accordance with the results obtained for the reactions with 12 and 13 (Table, entries 4 and 6), indicating mainly retention during the borylation. However, the differences in selectivity obtained for the reactions with 11 and 14 could be explained by either partial inversion during the borylation or the preferred reaction of one carbanion and partial nonproductive decomposition of the other. Deuteration of 34a provided a single diastereoisomer (dr ≥19:1). This result matches the selectivities obtained for 39a, 40a, and 42a (Table, entries 1, 3, and 7), providing evidence for full retention during the borylation of these substrates. The slightly lower selectivity obtained for 41a (entry 5) could be explained by partial inversion during this step. For Cb carbamate 34b, no selectivity was observed during deuteration (dr 1.1:1), which is in line with the result of the reaction with VBE 11 (entry 2). The differing selectivities obtained for all other substrates (entries 4, 6, and 8) could again be explained by partial inversion during the borylation or the preferred reaction of one carbanion and partial nonproductive decomposition of the other. To explain the observed very good to excellent selectivities obtained for the transformations of TIB ester 33a and 34a, we proposed a model based on minimization of steric repulsion and stabilizing electronic effects? (Scheme). For both TIB esters, we propose the deprotonation of the sterically less hindered proton. In the case of 33a, this would lead to Li-33a as the intermediate carbanion. As the deprotonation of 33a is not fully selective (dr 6:1), the diastereoisomer of Li-33a (not shown) is formed as well. In this, the carbanion would point toward the acetonide, which could explain a preference for inversion during the borylation step due to steric hindrance. In addition to this steric argument, orbital overlap could also stabilize conformer Li-33a. The C–Li σ-orbital overlaps with the σ*-orbital of the neighboring C–C bond, which in turn overlaps with the σ*-orbital of the neighboring O–C bond of the acetonide. Based on the same assumptions, the selectivities obtained for 34a could be explained with Li-34a as the carbanion. The close proximity of the methylene group that forms the carbanion and the geminal methyl groups of the acetonide might also explain the high level of diastereoselectivity during the borylation.
Proposed Transition States Rationalizing Our Observed Selectivities (σ-Orbitals Are Indicated by Red Dotted Lines)*
When we compare our results to the abovementioned work of Aggarwal et al.,? the huge differences in diastereoselectivity could also be explained with our model. In the twist-boat conformation of an anti-acetonide, steric interactions between the substituents might be less pronounced leading to a less defined transition state. Additionally, the stabilization by electronic effects is not given in a twist-boat conformation.
To summarize our studies on carbanions with two neighboring stereocenters (Figure), we differentiated our substrates by their conformational flexibility given by the used protecting groups first. For flexible substrates bearing a silyl ether as the protecting group, we obtained the formal Felkin products, when TIB esters were used as directing groups. In combination with large VBEs, excellent selectivities were obtained, which dropped to moderate, when small VBEs were applied. The selectivity for the reaction with small VBEs could be improved for anti-diketides, when coordinating protecting groups were used. However, this was only applicable to anti-diketides, since syn-diketides still provided moderate selectivities. For Cb carbamates, the formal anti-Felkin products were obtained in moderate selectivities regardless of the size of the VBEs. More rigid substrates bearing a six-membered acetonide as the protecting group provided the formal Felkin products in excellent selectivities for all applied TIB esters. When Cb carbamates were used, the selectivity dropped to moderate again, but the obtained major diastereoisomer did not always change to the formal anti-Felkin product.
Guidance flowchart for the substrate- and reagent-controlled lithiation–borylation chemistry of diketides and VBEs.
Following our detailed investigations on diketides bearing two consecutive stereocenters next to the directing group, we were interested in the effect of an additional third stereocenter. The idea that remote stereocenters might affect the stereochemical outcome of the reaction was based on previous studies on aldol reactions, showing the impact of the overall conformation of substrates on their outcome.? We started our investigations with acetonide-protected diketides,? as we estimated that the effect of distal stereocenters would be more pronounced in rigid systems due to potential syn-pentane interactions. The potential transition states of the acetonide-protected substrates with three contiguous stereocenters are shown below the corresponding table (Tables, ?, ?, and ?) and are based on minimization of syn-pentane interactions and removal of the less hindered proton as well as maximization of stabilizing electronic effects. A more detailed examination of these transition states is provided in our previous study.? We also decided to no longer apply four different vinyl boronic esters, but only VBEs 12 and 14, as the results for both branched VBEs 11 and 12 were usually comparable. Additionally, vinylboronic acid pinacol ester (14) was chosen as it showed the lowest selectivities so far. All-syn-diketides 43a/b (Table) provided the formal Felkin products as the major diastereoisomers. With TIB esters as directing groups (entries 1 and 2), excellent selectivities (dr 19:1) along with moderate to very good yields were obtained. In the case of Cb carbamate 43b and VBE 14 (entry 3), the formal Felkin product was obtained in a moderate selectivity of 3:1 along with a low yield.?
8: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of All-syn-Diketides 43a/b
9: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of anti, syn-Diketides 46a/b
10: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of syn, anti-diketides 49a/b
11: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of anti, anti-diketides 52a/b
The reactions of TIB ester 46a (anti, syn-diketide, Table, entries 1 and 3) with both VBEs led again to the formal Felkin products in excellent selectivities and very good yields. For its Cb counterpart 46b, the formal Felkin product 47a was obtained in a moderate selectivity and very good yield in the transformation with VBE 12 (entry 2). With unsubstituted vinylboronic acid pinacol ester (14) (entry 4), a slight preference toward the formal anti-Felkin product 48b was observed. This low level of selectivity was accompanied by a low yield.?
Surprisingly, nearly no selectivity was obtained for the reaction of syn, anti-TIB ester 49a and methyl-branched VBE 12 (Table, entry 1).? As the TIB esters of acetonide-protected diketides usually provided the higher level of diastereoselectivity with sterically demanding VBEs, we did not perform this transformation with its Cb analogue. In the reaction with 14, we obtained formal anti-Felkin product 51b as the major diastereoisomer regardless of the directing group (entries 2 and 5). Interestingly, TIB ester 49a provided a higher level of selectivity (dr 1:6) along with a much higher yield (72%) compared to Cb carbamate 49b (1:2, 14%). The selectivity for the reaction of 49a and 14 could be slightly increased to 1:7, when TMEDA was replaced by the (+)-sparteine surrogate (entry 4). It is noteworthy that no product formation was observed, when the bulkier (+)-sparteine was used as diamine ligand (entry 3), which further supports that the size of the ligand plays a crucial role for this transformation.?
Formal anti-Felkin product 53b was obtained as the major diastereoisomer in the reaction of methyl-branched VBE 12 and both anti, anti-diketides 52a/b (Table, entries 1 and 2). The anti, anti-configuration is a rare example where the Cb carbamate did provide a higher level of diastereoselectivity than the TIB ester (1:6 vs 1:2). However, the yield was higher for the reaction of TIB ester 52a (75% vs 60%). For the reaction of 52a with VBE 14 (entry 3), no diastereoisomer was favored (dr 1:1). Attempts to increase the diastereoselectivity by the use of sparteine (entries 4 and 5) failed due to no product formation. In contrast to these results, Cb carbamate 52b (entry 6) led to the formation of formal anti-Felkin product 54b in an excellent selectivity (1:19) but low yield.?
Taking these results as evidence that distal stereocenters affect the stereochemical outcome of this reaction, we next turned our attention to more flexible stereotriads bearing two silyl ethers. For all-syn-TIB ester 55a, a high level of diastereoselectivity was obtained in the reaction with branched VBE 12 (13:1, Table, entry 1) leading to formal Felkin product 56a. The same VBE provided no level of selectivity (dr 1:1), when Cb carbamate 55b was used (entry 2). With 14, both diketides led to formal anti-Felkin product 57b as the major diastereoisomer (entries 3 and 4). The yields of both reactions were comparable; however, Cb carbamate 57b provided the higher level of diastereoselectivity (1:3 vs 1:1.2). In accordance with previous results for more flexible Cb carbamates (Table, entries 11 and 12, and Table, entry 12), a full level of stereocontrol was obtained, when the reaction was performed in the presence of sparteine (entries 5 and 6).
12: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of All-syn-diketides 55a/b
In the reaction of anti, syn-TIB ester 58a and VBE 14, formal Felkin product 59a was obtained as the sole diastereoisomer (Table, entry 1). This is the second example for an excellent selectivity of an anti, syn-configured TIB ester, as this structural motif was also present in our synthesis of chondrochloren A (8) (Schemec).? Based on this observation and the excellent result for the reaction of 58a and 14, we did not investigate the combination of 58a and 12. The reaction of Cb carbamate 58b and 14 (entry 2) provided the formal anti-Felkin product in a slight excess (dr 1:1.2). Attempts to increase the selectivity of this reaction by the addition of sparteine (entries 3 and 4) failed due to no product formation.
13: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of anti, syn-Diketides 58a/b
Both syn, anti-diketides 60a/b provided moderate yields in the reaction with branched VBE 12 (Table, entries 1 and 2). For TIB ester 60a, formal Felkin product 61a was obtained in a good selectivity (dr 11:1). Its Cb counterpart 60b led to formal anti-Felkin product 61b in a good selectivity (dr 1:11) as well. Interestingly, TIB ester 60a and VBE 14 gave formal anti-Felkin product 62b in a low selectivity (dr 1:2, entry 3). The formal Felkin selectivity of the TIB ester was restored by the use of (−)-sparteine (entry 5), while in contrast to this result, the use of (+)-sparteine (entry 4) did not lead to any product formation. Formal anti-Felkin product 62b was obtained as a single diastereoisomer in the reaction of Cb carbamate 60b and VBE 14 (entry 6). The same level of diastereoselectivity along with a lower yield was obtained, when (+)-sparteine was used (entry 7), providing more evidence for the impact of the size of the diamine ligand on the yield of the reaction.
14: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of syn, anti-Diketides 60a/b
For the anti, anti-configuration (Table), formal Felkin product 64a was obtained in a good selectivity (dr 10:1), when TIB ester 63a was combined with 12 (entry 1). In combination with Cb carbamate 63b (entry 2), formal anti-Felkin product 64b was obtained in a low level of diastereoselectivity (dr 1:2). Simple vinylboronic acid pinacol ester (14) and TIB ester 63a (entry 3) provided formal Felkin product 65a in a slight excess (dr 2:1). The selectivity of this reaction could not be changed by the use of sparteine (entries 4 and 5) due to no product formation. In the reaction with Cb carbamate 63b (entry 6), the formal anti-Felkin product was obtained as a single diastereoisomer.
15: Substrate and Reagent Induction in Lithiation–Borylation Chemistry of anti, anti-Diketides 63a/b
Summarizing our work on TIB diketides bearing three contiguous stereocenters (Figure), we observed excellent formal Felkin selectivities for rigid substrates, when the stereocenters at C3 and C4 were syn-configured. These selectivities were obtained regardless of the size of the used vinyl boronic esters. However, with an anti-relation between C3 and C4, only moderate selectivities were obtained with both VBEs. Additionally, there was no general trend favoring either the formal Felkin or formal anti-Felkin product for these substrates. More flexible substrates provided excellent formal Felkin selectivities with large vinyl boronic esters regardless of the stereochemical relation between C3 and C4. For small VBEs, a moderate formal Felkin selectivity was obtained, when an anti-configuration between C3 and C4 was present. For a syn-relation between C3 and C4, no preference regarding the formation of either the formal Felkin or formal anti-Felkin product could be observed.
Guidance flowchart for the substrate- and reagent-controlled lithiation–borylation chemistry of TIB diketides bearing three contiguous stereocenters and VBEs.
In terms of Cb carbamates (Figure), a 3,4-syn-configuration in rigid substrates provided moderate selectivities for both large and small vinyl boronic esters and no general trend regarding the formation of the formal Felkin or formal anti-Felkin products. When an anti-relation was present, the formal anti-Felkin products were obtained in all cases. With large VBEs, moderate selectivities were obtained, whereas small reaction partners provided moderate to excellent selectivities. For more flexible substrates, a syn-configuration led to the formal anti-Felkin product in moderate selectivities with small VBEs. With larger reaction partners, no general preference toward one diastereoisomer was observed. In terms of an 3,4-anti-relation, the formal anti-Felkin products were formed in all reactions. For large VBEs, varying selectivities were obtained; however, with their small analogues, excellent diastereomeric ratios were obtained.
Guidance flowchart for the substrate- and reagent-controlled lithiation–borylation chemistry of Cb diketides bearing three contiguous stereocenters and VBEs.
Conclusions
In conclusion, we have demonstrated the potential of substrate induction in the stereoselective synthesis of allylic alcohols through lithiation–borylation chemistry. We showed that most of the applied TIB esters led to the formal Felkin products in excellent selectivities. Access to the formal anti-Felkin products could be obtained for most of the conformational flexible substrates, when the TIB group was replaced by the Cb group. This shows the potential for stereodivergent applications of this methodology even though the formal anti-Felkin selectivities were usually lower. Due to the good yields and selectivities obtained for more complex substrateswhich is also in line with our observations during the total synthesis of chondrochloren A (8)?we believe that this methodology will be applied extensively in the syntheses of natural products or other complex molecules in the future, especially as a valuable alternative to the NHTK reaction.
Experimental Section
Substrate-Controlled 1,2-Metallate Rearrangement of Vinyl Boronates
General Procedure for the Conversion of TIB Esters
To a stirred solution of TIB ester (1.5 equiv) and diamine (1.5 equiv) in Et_2_O (0.2 M) at −78 °C was added sBuLi (1.3 M in hexanes, 1.4 equiv). The reaction mixture was stirred for 5 h at that temperature before a solution of vinyl boronic ester (1.0 equiv) in Et_2_O (0.5 M) was added. After stirring for further 3 h at −78 °C, the reaction mixture was warmed to 45 °C and stirred overnight. The reaction mixture was cooled to rt, sat. aq. NH_4_Cl was added, and the biphasic mixture was stirred for 15 min. The phases were separated, the organic layer was washed with sat. aq. NH_4_Cl (3×) and the combined aqueous phases were extracted with MTBE (3×). The combined organic phases were dried over Na_2_SO_4_ and concentrated in vacuo and the crude material was purified by a short flash column chromatography (to remove TIBOH).? The residue was dissolved in THF (0.2 M) and cooled to −20 °C. A premixed, ice-cooled solution of NaOH (2.0 M)/H_2_O_2_ (35%, 2/1 v/v, 0.12 M) was added dropwise. The reaction mixture was stirred at rt before being diluted with MTBE and quenched by the slow addition of sat. aq. Na_2_S_2_O_3_ at 0 °C after TLC showed full conversion. The phases were separated, and the aqueous phase was extracted with MTBE (3×). The combined organic layers were dried over Na_2_SO_4_ and concentrated in vacuo. The crude product was purified by flash column chromatography to afford allylic alcohol.?
General Procedure for the Conversion of Carbamates
To a stirred solution of carbamate (1.5 equiv) and diamine (1.5 equiv) in Et_2_O (0.2 M) at −78 °C was added sBuLi (1.3 M in hexanes, 1.4 equiv). The reaction mixture was stirred for 5 h at that temperature before a solution of vinyl boronic ester (1.0 equiv) in Et_2_O (0.5 M) was added. The reaction mixture was stirred for 3 h at −78 °C.? In parallel, magnesium turnings were activated (2× 1.0 M HCl, 2× H_2_O, 2× acetone, drying under high vacuum). The required amount (2.0 equiv) was dissolved in Et_2_O (0.8 M), and 1,2-dibromoethane (2.0 equiv) was added under water bath cooling. The reaction mixture was stirred for 2 h at this temperature.? The biphasic MgBr_2_·OEt_2_ solution was added dropwise to the main reaction mixture, which was then stirred for another 30 min at −78 °C before being warmed to 45 °C and stirred overnight. The reaction mixture was cooled to rt, sat. aq. NH_4_Cl was added, and the biphasic mixture was stirred for 15 min. The phases were separated, the organic layer was washed with sat. aq. NH_4_Cl (3×), and the combined aqueous phases were extracted with MTBE (3×). The combined organic phases were dried over Na_2_SO_4_ and concentrated in vacuo, and the crude material was purified by a short flash column chromatography (to remove excess of the carbamate).? The residue was dissolved in THF (0.2 M) and cooled to −20 °C. A premixed, ice-cooled solution of NaOH (2.0 M)/H_2_O_2_ (35%, 2/1 v/v, 0.12 M) was added dropwise. The reaction mixture was stirred at rt before being diluted with MTBE and quenched by the slow addition of sat. aq. Na_2_S_2_O_3_ at 0 °C after TLC showed full conversion. The phases were separated, and the aqueous phase was extracted with MTBE (3×). The combined organic layers were dried over Na_2_SO_4_ and concentrated in vacuo. The crude product was purified by flash column chromatography to afford allylic alcohol.?
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Rodríguez J.Riguera R.New Marine Cytotoxic Bispyrones. Absolute Stereochemistry of Onchitriols I and II Tetrahedron Lett.1992331089109210.1016/S 0040-4039(00)91868-9 · doi ↗
- 2a Okude Y.Hirano S.Hiyama T.Nozaki H.Grignard-type carbonyl addition of allyl halides by means of chromous salt. A chemospecific synthesis of homoallyl alcohols J. Am. Chem. Soc.1977993179318110.1021/ja 00451 a 061 · doi ↗
- 3a Kobayashi K.Fujii Y.Hirayama Y.Kobayashi S.Hayakawa I.Kigoshi H.Design, Synthesis, and Biological Evaluations of Aplyronine A–Mycalolide B Hybrid Compound Org Lett.2012141290129310.1021/ol 300182 r 22356580 · doi ↗ · pubmed ↗
- 4a Leonori D.Aggarwal V. K.Lithiation–Borylation Methodology and Ist Application in Synthesis Acc. Chem. Res.2014473174318310.1021/ar 500247325262745 · doi ↗ · pubmed ↗
- 5Linne Y.Lohrberg D.Struwe H.Linne E.Stohwasser A.Kalesse M.1,2-Metallate Rearrangement as a Toolbox for the Synthesis of Allylic Alcohols J. Org. Chem.202388126231262910.1021/acs.joc.3c 0130937594929 PMC 10476192 · doi ↗ · pubmed ↗
- 6Linne Y.Bonandi E.Tabet C.Geldsetzer J.Kalesse M.The Total Synthesis of Chondrochloren A Angew Chem. Int. Ed.2021606938694210.1002/anie.202016072 PMC 804895833450788 · doi ↗ · pubmed ↗
- 7a Linne Y.Birkner M.Flormann J.Lücke D.Becker J. A.Kalesse M.Sparteine-Free, Highly Stereoselective Construction of Complex Allylic Alcohols Using 1,2-Metallate Rearrangements JACS Au 202331695171010.1021/jacsau.3c 0011437388702 PMC 10301690 · doi ↗ · pubmed ↗
- 8Ritter S. K.Where has all the sparteine gone?C&EN 201795182010.1021/cen-09517-scitech 1 · doi ↗