Exhaustion‐Resistant CD8 + T Cells in Ankylosing Spondylitis: A Proposed Three‐Axis Model
Xuhong Zhang, Lu Jia, Xueni Lin, Lamei Zhou

TL;DR
The paper explores how certain CD8+ T cells in ankylosing spondylitis resist exhaustion and contribute to chronic inflammation and bone changes.
Contribution
It introduces a novel three-axis model explaining the persistence of functional CD8+ T cells in ankylosing spondylitis.
Findings
Exhaustion-resistant CD8+ T cells retain effector function and contribute to tissue inflammation and bone remodeling.
A three-axis model involving TCR stimulation, IL-15/CD28 co-stimulation, and metabolic fitness explains CD8+ T cell persistence.
Potential biomarkers and therapeutic strategies, such as JAK–IL-15 pathway modulation, are proposed.
Abstract
Ankylosing spondylitis (AS) is a chronic immune‐mediated disease marked by sustained joint inflammation and aberrant bone remodelling. Although chronic antigen exposure usually enforces terminal exhaustion, emerging evidence indicates that a subset of CD8+ T cells in AS evades canonical exhaustion programmes while expressing programmed cell death protein 1 (PD‐1). These exhaustion‐resistant cells retain effector function and likely contribute to persistent tissue inflammation and structural damage. In this review, we dissect the cellular and molecular basis of exhaustion resistance in AS CD8+ T cells and focus on the convergence of intermittent T cell receptor (TCR) stimulation, metabolic adaptation that preserves mitochondrial fitness, and co‐stimulatory inputs from interleukin‐15 (IL‐15) and CD28. We propose an integrated three‐axis model governing CD8+ T cell fate and functional…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
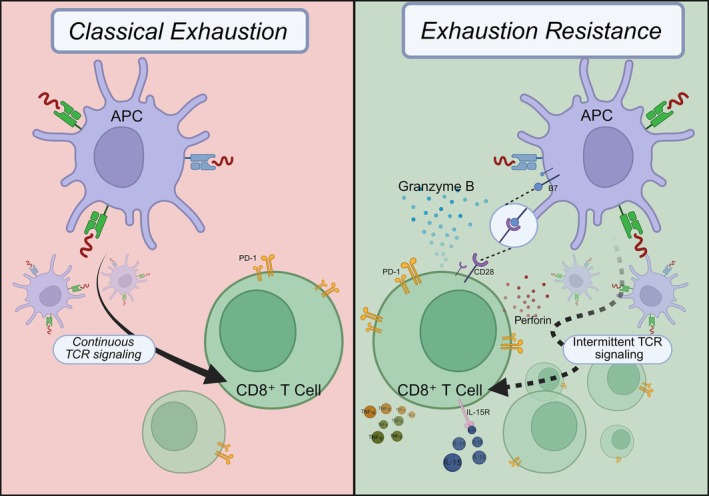 FIGURE 1
FIGURE 1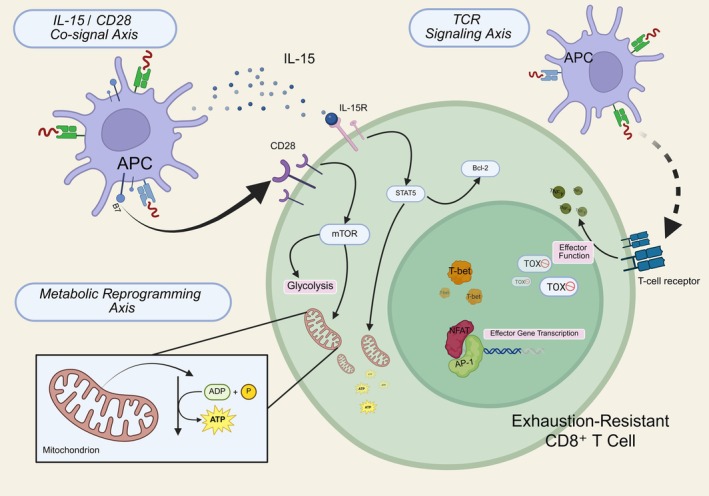 FIGURE 2
FIGURE 2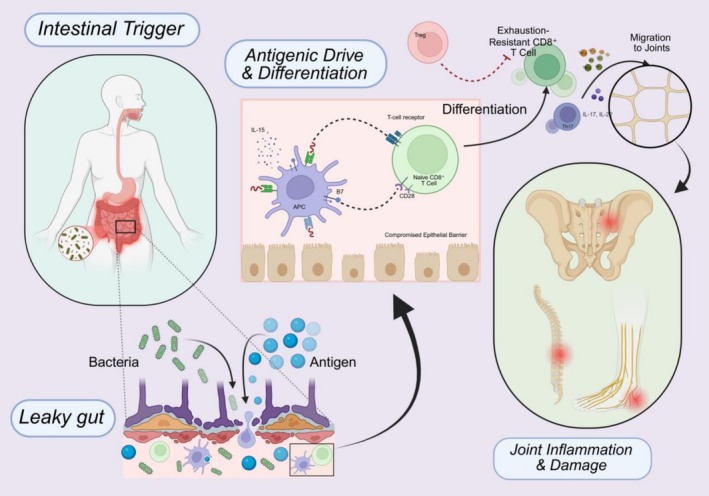 FIGURE 3
FIGURE 3- —Wuxi Municipal Health Commission, Wuxi Taihu Lake Talent Plan, Supports for Leading Talents in Medical and Health Profession
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmune Cell Function and Interaction · T-cell and B-cell Immunology · IL-33, ST2, and ILC Pathways
Introduction
1
Ankylosing spondylitis is an immune‐mediated inflammatory disease of the axial skeleton characterised by sacroiliac and spinal inflammation, fibrosis and osteoproliferation that can culminate in spinal fusion [1]. The disease typically begins in young adults and shows a strong genetic association with HLA‐B27 [2], which supports a central role for CD8^+^ T cells [3]. Beyond classical antigen presentation, HLA‐B27‐related phenomena, including misfolding‐induced unfolded protein response and interactions with killer cell immunoglobulin‐like receptors, link innate and adaptive compartments and help explain persistent inflammation [4, 5]. Biologics targeting tumour necrosis factor‐alpha (TNF‐α) or interleukin‐17 (IL‐17) improve symptoms but often fail to induce durable remission, indicating unresolved mechanisms [6, 7].
A consistent observation in AS is the presence of PD‐1^high CD8^+^ T cells that remain metabolically competent and cytotoxic [8, 9]. In chronic antigenic stimulation, CD8^+^ T cells may enter a state termed T‐cell exhaustion, characterised by progressive loss of effector function together with defined transcriptional and epigenetic programmes [10]. Rather than representing canonical exhaustion, these cells appear to resist the TOX‐driven programme and undergo clonal expansion at inflamed sites [11, 12], which is consistent with intermittent stimulation by self or microbial antigens [13]. This central paradox is visually summarised in Figure 1.
The paradox of CD8+ T‐cell exhaustion resistance in AS. Left: Classical CD8+ T‐cell exhaustion driven by continuous TCR signalling and characterised by progressive functional decline. Right: The exhaustion‐resistant phenotype observed in AS. In AS, intermittent antigenic stimulation together with CD28 co‐stimulation and interleukin‐15 allows CD8+ T cells to retain robust effector function and proliferative capacity despite the expression of inhibitory receptors such as PD‐1. (Created with BioRender.com).
Several AS‐specific features in AS help explain why this phenotype emerges and why our model is applicable. HLA‐B27 biology together with the ‘enthesis organ’ architecture creates niches for local immune activation; human enthesis contains resident lymphocytes that respond to interleukin‐23 and γδ T cells capable of interleukin‐17 production, supporting site‐restricted stimulation [14, 15, 16]. In parallel, a gut–joint axis is evident: subclinical intestinal inflammation is frequent in spondyloarthritis and can be tracked by faecal calprotectin, and an AS‐associated ileal microbial signature has been described [17, 18, 19]. Repetitive biomechanical stress at entheses provides episodic danger cues and antigen release, favouring intermittent and tissue‐confined TCR engagement rather than uniform, chronic exposure [20].
Against this backdrop, we advance a three‐axis framework that comprises intermittent TCR engagement, sustained metabolic fitness and IL‐15/CD28 co‐stimulation to account for exhaustion resistance and to generate testable predictions. We then integrate this framework with established AS pathways, including the interleukin‐23 (IL‐23)/IL‐17 circuit, ILC3 activity and gut barrier dysfunction, and we outline translational implications.
Classical Model of CD8
- T‐Cell Exhaustion
2
In acute infections, most effector CD8^+^ T cells undergo apoptosis after antigen clearance, with only a few differentiating into memory cells [21]. In chronic infections or cancer, however, continuous antigen exposure prevents CD8^+^ T cells from entering quiescence or forming long‐lived memory, instead driving them into a functionally constrained ‘exhausted’ state [22, 23]. Classically exhausted CD8^+^ T cells exhibit a characteristic phenotype and functional impairment: they co‐express multiple inhibitory receptors (e.g., PD‐1, CTLA‐4, LAG‐3, TIM‐3) [24], downregulate memory‐associated molecules, like IL‐7Rα (CD127) and CD62L, show reduced proliferative capacity, and undergo a hierarchical loss of effector functions (sequentially losing the ability to produce IL‐2, TNF‐α, then IFN‐γ) [25, 26]. At the transcriptional and epigenetic level, exhausted T cells establish a programme distinct from effector or memory T cells [27], governed by key regulators, such as T‐bet, Eomes, the NR4A family and TOX, among others, which collectively drive and maintain the exhausted cell fate. For example, the transcription factor TOX has been identified as essential for inducing and reinforcing the exhaustion programme—in its absence, CD8^+^ T cells in chronic infection fail to establish a stable exhausted phenotype [28]. These molecular mechanisms ensure the relative irreversibility of exhaustion, tightly regulating T‐cell responses under prolonged antigenic stimulation. Mechanistically, sustained TCR engagement in chronic settings activates the calcineurin–nuclear factor of activated T cells (NFAT) pathway in an altered mode (often unopposed by activator protein‐1 (AP‐1)), biasing gene expression towards an exhaustion programme. NFAT thereby induces transcription factors like TOX and NR4A that execute and lock in the exhaustion state [29, 30]. TOX in particular drives extensive epigenomic remodelling of T cells, cementing the exhausted phenotype. Consistently, without TOX, chronically stimulated CD8^+^ T cells show markedly reduced PD‐1 upregulation and retention of greater effector function despite persistent antigen exposure, underscoring TOX's central role in stabilising exhaustion.
From an evolutionary perspective, T‐cell exhaustion is viewed as a ‘necessary compromise’ – a trade‐off that sacrifices some pathogen‐clearing efficacy in chronic infection or cancer in exchange for protection of the host's own tissues [23]. By limiting T‐cell–mediated tissue damage, exhaustion is protective: for instance, in chronic viral infection models, the absence of negative regulators like PD‐1 leads to lethal immunopathology due to unchecked T cell activity [31]. Similarly, in autoimmunity, the exhaustion programme helps prevent self‐reactive T cells from inflicting rampant damage on host tissues [32]. The cost of this protection is attenuated immunity—exhausted CD8^+^ T cells are less able to eliminate persistent antigens (such as chronic HBV/HCV or tumour antigens), allowing pathogen persistence or tumour progression [27]. Recent studies further reveal that the exhausted T cell pool is heterogeneous, comprising subsets at different differentiation stages [33, 34]. Among these, progenitor or ‘stem‐like’ exhausted T cells (typically TCF‐1^+^ PD‐1^+^) retain some proliferative and differentiative capacity; they can self‐renew and give rise to partially re‐functionalised progeny, and they are the main subset reinvigorated by therapies like PD‐1 checkpoint blockade [35]. In contrast, terminally exhausted T cells (e.g., TIM‐3^+^ PD‐1^+^ TOX^high cells) are deeply dysfunctional, with minimal proliferative potential and resistance to reactivation [28]. In sum, the classical model describes a state of CD8^+^ T cells under chronic antigenic stress that is constrained by multiple inhibitory signals and locked in by transcriptional regulators. This state dampens T cell effector functions to strike a balance between pathogen clearance and prevention of host tissue damage.
Non‐Classically Exhausted CD8
- T Cells in AS
3
In this review, we use ‘exhaustion‐resistant’ to denote CD8^+^ T cells that (i) express one or more checkpoint receptors (most commonly PD‐1) and (ii) fail to engage the canonical TOX‐driven exhaustion programme (low or absent TOX with preserved TCF7/effector modules) and (iii) retain cytotoxic function and proliferative potential. Consistent with prior work, PD‐1 expression alone is not sufficient to infer exhaustion or exhaustion resistance.
Although T‐cell exhaustion is well‐established in infections and cancer, recent evidence indicates that atypical CD8^+^ T cell states exist in chronic inflammatory diseases, including autoimmune arthritis [36]. These T cells express certain ‘exhaustion’ markers on the surface but do not exhibit functional impairment; instead, they remain immunologically active. In juvenile idiopathic arthritis (JIA), for example, Petrelli et al. reported an accumulation of PD‐1^+^ CD8^+^ T cells in synovial fluid [36]; transcriptomic and functional assays showed that these cells were metabolically active cytotoxic effectors, but TOX was not assessed in that data set. We therefore do not classify these cells as exhaustion‐resistant by our definition; rather, they illustrate that checkpoint expression can occur outside terminal exhaustion in inflamed joints. Notably, similar PD‐1^+^ CD8^+^ subsets have been observed in lesional tissues of other chronic inflammatory diseases [34], again without uniform evidence for engagement of the TOX‐driven programme. In this review we reserve the term ‘exhaustion‐resistant’ for CD8^+^ T cells that co‐express checkpoint receptors yet show low or absent TOX together with retained effector function (see AS data sets cited herein) [24]. By contrast, in ankylosing spondylitis, we and others observe PD‐1^high CD8^+^ T cells with low or absent TOX and retained effector function, including clonal expansion at lesion sites, which satisfies our operational criteria for exhaustion resistance [8, 11, 12].
Studies in AS have reported analogous findings. In the peripheral blood and joint compartments of active AS patients, a distinct subset of CD8^+^ cytotoxic T lymphocytes (CTLs) has been identified that co‐expresses high levels of checkpoint receptors PD‐1, TIGIT and LAG‐3, yet still retains hallmark CTL functional capacities [8]. In a multi‐omics single‐cell analysis, Tang et al. found that PD‐1^+^ TIGIT^+^ CD8^+^ T cells in AS synovial fluid markedly downregulated the IL‐7 receptor α‐chain (CD127) but, upon stimulation, could still produce substantial IFN‐γ and TNF‐α and expressed cytolytic molecules like granzyme B and perforin. These cells also highly expressed tissue‐residency markers, such as CD103 and CD69, indicating their localisation at inflammatory sites [37]. Emerging evidence suggests a potential intrinsic link between TRM programmes and exhaustion‐resistance mechanisms, where TRM‐associated transcriptional networks (such as Hobit and Blimp‐1) may concurrently enhance tissue residency and limit exhaustion programming. Further elucidation of this link could substantially refine our understanding of CD8^+^ T‐cell persistence in chronic inflammation [38]. Crucially, single‐cell RNA sequencing revealed that this subset downregulated exhaustion‐associated genes and, most notably, the master transcription factor TOX, which is essential for establishing the canonically exhausted state in chronic infection or cancer [28]. The failure to upregulate TOX, possibly due to a distinct pattern of NFAT‐dominant signalling in the absence of AP‐1 in the AS microenvironment, likely prevents the stable epigenetic remodelling required for deep exhaustion, allowing these cells to remain in a highly functional state. Flow cytometry confirmed that these PD‐1^high CTLs were largely TOX^−^. Taken together, these findings support the existence of an exhaustion‐resistant CD8^+^ T‐cell subset in AS, in which inhibitory receptor expression coexists with preserved effector programmes (e.g., IFN‐γ, perforin). The apparent failure of the PD‐1 checkpoint to impose terminal exhaustion is more likely attributable to overriding pro‐inflammatory signals in the tissue microenvironment, as discussed below.
Notably, these exhaustion‐resistant CD8^+^ T cells tend to accumulate in lesional tissue (e.g., within the synovium of AS joints), in contrast to classically exhausted T cells that are often enriched in secondary lymphoid organs. The presence of such cells offers a potential explanation for the chronicity of AS: a subset of pathogenic CD8^+^ T cells may evade the canonical exhaustion programme and remain persistently aggressive against autoantigens, thereby driving prolonged inflammation and tissue damage. This concept shifts the paradigm beyond the previously dominant focus on the Th17/IL‐17 axis [39] and motivates renewed attention to the role of CD8^+^ T cells in AS immunopathology.
Potential Molecular Mechanisms: TCR Axis, Metabolic Axis and IL‐15/CD28 Axis
4
Why do certain CD8^+^ T cells in AS evade classical exhaustion? Current hypotheses centre on the combined influence of three factors: TCR signalling, cellular metabolic status and the co‐stimulatory/cytokine milieu. In this section, we discuss how each of these three pathways may modulate T‐cell exhaustion versus functional maintenance, and we propose how their interplay could operate in AS.
TCR Signalling Axis
4.1
The intensity and pattern of TCR signalling under chronic antigen exposure is a primary determinant of T‐cell fate. In general, continuous high‐intensity TCR stimulation drives the initiation of the exhaustion programme, but the intermittency and site‐specificity of stimulation can alter this outcome [28]. In AS, the autoantigens triggering inflammation may not be uniformly present continuously; rather, they are closely linked to biomechanical stress and the gut microbiome. Biomechanical stress at the entheses not only generates local damage‐associated molecular patterns (DAMPs) that stimulate resident T cells, but has been shown in HLA‐B27 transgenic rats to amplify inflammation via antigen release under mechanical loading, thereby reinforcing intermittent TCR activation [40]. Meanwhile, certain gut microbial antigens presented by HLA‐B27 may mimic self‐peptides, making gut inflammation an ‘engine’ that fuels systemic immune responses. For example, recent findings show that expanded CD8^+^ T cell clones in AS often use the TRBV9 gene and can recognise HLA‐B27—presented peptides from microbes and self, implicating antigenic drive along a gut–joint axis [41, 42]. However, unlike in chronic viral infections where antigen persists continuously, antigenic stimulation in AS may be episodic. It is hypothesised that flares triggered by gut microbiota changes or mechanical injury create cycles of recurrent stimulation and brief respite. According to a model primarily derived from chronic viral infection studies, such intermittent antigen exposure mitigates deep T‐cell exhaustion [26]. In this context, AS‐pathogenic CD8^+^ T cells might avoid the unrelenting signal overload that locks cells into an exhausted fate. This remains a key hypothesis for AS, as direct validation through methods like antigen‐specific TCR tracking in relevant models is still needed. Intermittent antigen exposure might allow epigenetic resetting or memory differentiation, thereby avoiding sustained NFAT‐driven TOX induction [43]. Concurrently, during each flare, abundant local antigen‐presenting cells likely provide strong co‐stimulatory signals, activating T cells in an effector mode rather than routing them into dysfunction.
Metabolic Reprogramming Axis
4.2
The metabolic state of a T cell is a critical intrinsic factor determining its function and fate. Exhausted T cells typically exhibit metabolic dysfunction: reduced mitochondrial mass and function, impaired oxidative phosphorylation, and an over‐reliance on glycolysis with diminished energetic efficiency [44]. In contrast, T‐cell subsets that are exhaustion‐resistant tend to maintain better mitochondrial function and metabolic flexibility. For example, progenitor ‘stem‐like’ exhausted T cells sustain higher mitochondrial respiration and fatty acid oxidation, which is thought to support their longevity and self‐renewal capacity [33, 45]. In inflammatory settings, exhaustion‐resistant CD8^+^ T cells appear to exhibit a highly active metabolic profile [46]. For example, PD‐1^+^ CD8^+^ T cells in JIA synovial fluid were found to have elevated glycolytic and mitochondrial activity to meet the demands of continuous proliferation and effector function [36]. However, it must be stressed that this is an extrapolation, as direct metabolic profiling of AS synovial T cells is currently a critical knowledge gap. We hypothesise that AS‐resident CD8^+^ T cells share this metabolic robustness, a point that urgently requires validation through techniques like Seahorse metabolic flux analysis and targeted metabolomics. On the other hand, the metabolic conditions of the tissue microenvironment (such as availability of oxygen, glucose and other nutrients) also influence T‐cell fate [47]. Although chronic inflammation in the synovium entails some degree of hypoxia and nutrient competition, it may not be as extremely suppressive as the tumour microenvironment [48]. Importantly, certain factors in chronic inflammation can activate metabolism‐favouring pathways; for instance, pro‐inflammatory cytokines can enhance T cell glucose uptake and glycolysis via mTOR signalling, thereby sustaining effector functions. Notably, PD‐1 ligation attenuates the phosphoinositide 3‐kinase–AKT (protein kinase B)–mechanistic target of rapamycin (PI3K–AKT–mTOR) signalling pathway in T cells, downregulating glucose transporter and glycolytic enzyme expression and shifting metabolism towards fatty acid oxidation [49, 50]. In exhaustion‐prone settings, this metabolic brake limits T‐cell effector capacity [51]. However, the unexhausted PD‐1^high CD8^+^ T cells in AS likely bypass this brake—Concurrent inflammatory signals, particularly from IL‐15 and CD28‐mediated co‐stimulation, appear to sustain Akt/mTOR signalling despite high PD‐1 expression, thus preserving glucose uptake capacity and mitochondrial functionality, although direct evidence from AS‐specific analyses is warranted [52]. This metabolic robustness of PD‐1^+^ CD8^+^ effectors in AS mirrors similar observations in chronic arthritis, suggesting a preserved effector phenotype despite inhibitory signalling. Of course, there is a flip side: excessive mTOR activation and glycolytic bias might accelerate exhaustion, as studies show that mitochondrial dysfunction coupled with HIF‐1α–driven glycolytic reprogramming can hasten the transition of precursor exhausted T cells into a terminally exhausted state [45, 53]. Therefore, in AS, exhaustion‐resistant CD8^+^ T cells maintain an optimal metabolic equilibrium characterised by mitochondrial flexibility and balanced glycolytic flux—possessing sufficient fuel and bioenergetic support to maintain cytotoxic activity, yet avoiding the mitochondrial collapse that triggers irreversible dysfunction. Cytokines like IL‐7 and IL‐15 may aid in this balance by promoting mitochondrial homeostasis and metabolic fitness in memory/resident T cells.
IL‐15/CD28 Co‐Stimulation Axis
4.3
Beyond the TCR and metabolic factors, the cytokine and co‐stimulatory milieu is pivotal in steering T cell functional states. In inflamed AS joints, innate immune and stromal cells produce abundant common γ‐chain cytokines (like IL‐7, IL‐15) and express co‐stimulatory ligands (like CD80/86), providing ‘signal 3’ cytokine support and co‐stimulation to T cells. IL‐15, in particular, is homologous to IL‐2 but more geared towards supporting memory and resident T‐cell survival. IL‐15 can promote CD8^+^ T‐cell survival and proliferation via signal transducer and activator of transcription 5 (STAT5) and mTOR pathways without inducing overt exhaustion or activation‐induced cell death [54]. In a chronic infection mouse model, exogenous IL‐15 selectively expanded PD‐1^+^ TCF‐1^+^ progenitor exhausted cells far more than terminally exhausted cells, thereby enhancing the self‐renewal and maintenance of the former [55]. A similar effect was observed in human tumour‐infiltrating lymphocytes: IL‐15 preferentially expanded a stem‐like subset of PD‐1^+^ CD8^+^ T cells [56]. These findings suggest that in AS inflamed joints, abundant IL‐15 signalling might prevent CD8^+^ T cells from prematurely exhausting, sustaining them in a more memory‐like, functional state. Meanwhile, ongoing CD28/B7 co‐stimulation is another crucial factor for preserving T cell function. PD‐1's inhibitory effect is largely executed by blunting CD28 signalling; therefore, if antigen‐presenting cells (APCs) in the inflammatory milieu continuously deliver CD28 co‐stimulation, the suppressive impact of PD‐1 signalling is partly offset, allowing T cells to retain a degree of effector activity [52]. Mechanistically, PD‐1 recruits SHP‐2 phosphatase to dephosphorylate CD28 and its downstream signalling components, dampening PI3K/Akt activation [49]. Continuous B7:CD28 engagement can thus counteract PD‐1–mediated signalling blockade, preserving Akt/mTOR‐driven functionality of the T cell [57]. Indeed, chronic infection models show that absence of CD28 co‐stimulation causes accelerated T‐cell dysfunction [10], whereas sustained low‐level CD28 signals favour long‐term maintenance of antiviral T cells. In AS patients, although a CD28^−^ exhausted phenotype subset exists in circulation, the activated CTLs at the joint often still express co‐stimulatory molecules and receive helper signals [58, 59]. Furthermore, the influx of myeloid cells and dendritic cells in AS synovium provides robust B7:CD28 co‐stimulation [60, 61]. Collectively, the IL‐15 and CD28 axes constitute a critical counter‐regulatory network: IL‐15 sustains T‐cell survival through STAT5‐Bcl2 signalling, while CD28 co‐stimulation preserves PI3K‐mTOR activity, synergistically promoting T‐cell persistence and actively antagonising inhibitory signals imposed by markers like PD‐1, thereby shaping the non‐exhausted, high‐functionality state of CD8^+^ T cells in AS [55, 62].
Integration of the Three Pathways: A Unified Model
5
We propose a mechanistically integrated model in which the exhaustion‐resistant phenotype of CD8^+^ T cells in AS emerges from the coordinated influence of three axes: persistent but intermittent TCR engagement, metabolic resilience and compensatory co‐stimulation through IL‐15 and CD28 (Figure 2). This triad forms a regulatory equilibrium between pro‐exhaustion cues and functional maintenance signals.
Integrated three‐axis model supporting exhaustion resistance. A mechanistic model illustrating how three interlinked axes synergise to preserve CD8+ T cell functionality in AS: (i) the IL‐15/CD28 co‐stimulation axis promotes survival and metabolic adaptation through STAT5 and mTOR signalling; (ii) the TCR signalling axis shows how intermittent stimulation supports effector gene transcription while preventing TOX‐mediated terminal exhaustion; (iii) the metabolic reprogramming axis highlights sustained mitochondrial fitness and glycolytic flexibility. (Created with BioRender.com).
First, chronic but episodic antigenic stimulation—arising from biomechanical stress or gut‐derived antigens [63]—induces expression of inhibitory receptors like PD‐1, priming T cells towards an exhaustion trajectory [23]. However, in contrast to tumours or persistent viral infections, antigenic exposure in AS may be spatially and temporally confined, allowing partial reset of exhaustion programmes and epigenetic plasticity between flares.
Second, metabolic adaptability serves as a critical intracellular buffer. AS‐associated CD8^+^ T cells retain mitochondrial fitness and maintain moderate glycolytic flux, enabling sustained cytotoxic function while avoiding bioenergetic collapse [64, 65].
Third, the inflammatory synovial milieu in AS provides abundant CD28 co‐stimulation and common gamma‐chain (γc) cytokines such as IL‐15, which counterbalance PD‐1–mediated suppression by sustaining PI3K‐Akt–mTOR activity and upregulating pro‐survival programmes such as Bcl‐2 via STAT5. This extrinsic reinforcement promotes cellular persistence without functional attrition.
When these anti‐exhaustion signals collectively prevail, CD8^+^ T cells assume a ‘para‐exhausted’ or ‘functionally unexhausted’ state—characterised by surface expression of inhibitory receptors but lacking TOX‐driven epigenetic reprogramming. These cells maintain robust cytotoxicity, effector function and clonal persistence, thereby serving as long‐term contributors to inflammation and tissue injury in AS.
This model generates testable hypotheses: modulating the strength or synchronisation of these axes—e.g., reducing IL‐15 availability, inhibiting CD28 signalling or disrupting mitochondrial integrity—may shift CD8^+^ T cells towards terminal exhaustion and reduce pathogenicity. Such strategies may support future therapeutic designs that reprogram exhaustion resistance in AS‐specific immune circuits.
Integration With Other Immune Pathways in AS
6
Placing our model of exhaustion‐resistant CD8^+^ T cells into the broader context of AS pathogenesis, we must consider its interplay with other well‐known immune pathways in the disease. AS is understood as a coordinated outcome of multiple cellular and molecular pathways, including an imbalance between T helper 17 (Th17) cells and regulatory T cells (Tregs) [66], an IL‐23/IL‐17 axis in which type 3 innate lymphoid cells (ILC3) are key contributors [67], and dysregulation of the gut microbiome together with impairment of the intestinal epithelial and vascular barriers [68, 69]. The presence of highly functional, exhaustion‐resistant CD8^+^ T cells adds a new dimension to this network and intersects with other pathways in several ways [8, 9]. This complex interplay, from the intestinal trigger to the multi‐cellular interactions within the joint microenvironment leading to pathology, is summarised in Figure 3.
Role of exhaustion‐resistant CD8+ T cells in the gut–joint axis of AS. A schematic overview of AS pathogenesis centered on exhaustion‐resistant CD8+ T cells. Intestinal trigger: Disruption of the gut barrier permits microbial translocation. Antigenic drive and differentiation: Microbial and self‐antigens activate naive CD8+ T cells through antigen‐presenting cells (APCs) with interleukin‐15 (IL‐15)/CD28 co‐stimulation, promoting differentiation towards an exhaustion‐resistant phenotype. Migration to joints: These cells circulate and accumulate at inflamed entheses. Joint osteoimmune microenvironment: Exhaustion‐resistant CD8+ T cells contribute to inflammation and bone remodelling together with T helper 17 (Th17) cells, type 3 innate lymphoid cells (ILC3) and regulatory T cells (Tregs). (Created with BioRender.com.)
Th17/IL‐23/IL‐17 Axis
6.1
A central pathogenic pathway in AS is the IL‐23–driven axis of Th17 cells and ILC3 producing IL‐17. IL‐17 and related cytokines (such as IL‐22) are crucial in joint inflammation and bone remodelling: IL‐17 promotes local inflammation and contributes to bone erosion [70], whereas IL‐22 is implicated in osteoproliferation [71]. These cytokines may disrupt the balance between bone resorption and formation, thereby driving both osteolysis and syndesmophyte formation—hallmarks of the paradoxical osteoimmune phenotype in AS. By secreting IFN‐γ, TNF‐α and IL‐22, exhaustion‐resistant CD8^+^ T cells in AS contribute to both osteoclastogenesis and enthesis ossification through the RANKL and BMP/Smad pathways. Separately, IL‐17–producing CD8^+^ T cells (Tc17) have also been described in AS. Whether these exhaustion‐resistant CD8^+^ T cells phenotypically or functionally overlap with the Tc17 population remains unresolved and represents a key direction for future investigation [72]. Future studies employing single‐cell multi‐omics analyses to co‐profile exhaustion markers (e.g., TOX), cytokine production (e.g., IL‐17, IFN‐γ) and TCR clonality in AS synovial fluid could elucidate this potential overlap. Meanwhile, IFN‐γ activates macrophages and synoviocytes to release additional chemokines and MMPs, amplifying inflammation and facilitating Th17 cell recruitment; TNF‐α synergises with IL‐17, jointly mediating inflammation and tissue destruction, which is why combined blockade of these cytokines yields enhanced clinical efficacy [73]. Moreover, IL‐17 can reciprocally affect CD8^+^ T cell activity; for example, IL‐17–driven inflammation can mature APCs and upregulate co‐stimulatory molecules, thereby boosting CD8^+^ T‐cell activation [74]. In sum, CD8^+^ T‐cell exhaustion‐resistance and the Th17/IL‐17 axis are not isolated—they mutually reinforce each other, acting as dual engines of chronic inflammation.
Treg Dysfunction
6.2
Regulatory T cells (Tregs) are crucial for maintaining immune tolerance, but their suppressive function appears compromised in AS [75]. While some studies report increased Treg proportions, these cells fail to suppress pathogenic T cells effectively [76]. Exhaustion‐resistant CD8^+^ T cells may explain this paradox, as they likely exhibit reduced sensitivity to Treg‐mediated suppression [77]. This resistance could be multifactorial. First, the strong pro‐survival and pro‐activation signals these cells receive, such as from IL‐15 via the STAT5 pathway, may make them inherently more resistant to suppressive signals. Second, the high levels of IFN‐γ they secrete can directly impair Treg function or render effector T cells less susceptible to Treg inhibition. Finally, while Tregs can use the PD‐1/PD‐L1 axis for suppression [78], this pathway is functionally antagonised in exhaustion‐resistant cells, blunting a key mechanism of Treg control. This creates a self‐reinforcing pathogenic circuit where dysfunctional Tregs fail to restrain hyperactive CD8^+^ T cells, whose inflammatory products in turn further limit Treg efficacy.
ILC3 and the Gut Axis
6.3
Innate lymphoid cells, especially ILC3, play a key role in the gut–joint axis of AS. ILC3 produce IL‐17 and IL‐22 in the gut mucosa and joints in response to IL‐23 [79]. HLA‐B27 transgenic rat studies show that a germ‐free environment prevents SpA‐like disease, whereas reintroduction of gut microbes triggers intestinal inflammation followed by arthritis [80]. This underscores the initiating role of the gut microbiome and barrier in AS. The CD8^+^ T‐cell exhaustion‐resistance concept complements this axis: the gut microbiota provides chronic but submaximal antigenic stimulation, activating certain clones of CD8^+^ T cells which then migrate to the joints. There, these T cells cooperate with cells like ILC3 to shape the inflammatory milieu. IL‐17/IL‐22 from ILC3 act on stromal cells to induce inflammatory mediators and drive bone changes, while IFN‐γ and TNF‐α from CD8^+^ T cells amplify this cytokine network. Conversely, cytokines from CD8^+^ T cells can affect barrier function: for example, IFN‐γ can increase gut epithelial permeability or alter microbiota composition, creating a ‘leaky gut’ and translocation of microbes that provide further T cell stimuli. This could explain why a significant fraction of AS patients have subclinical gut inflammation even without overt IBD.^19^ In essence, the persistence and hyperactivity conferred by the exhaustion‐resistant phenotype allow these CD8^+^ T cells to effectively link the ILC3–gut axis with the adaptive immune axis, forming a pivotal node in the pathogenic network of AS.
Testing the Proposed Model: An Experimental Roadmap
7
Samples and Comparators
7.1
Prioritise lesion‐site material whenever feasible, including synovial fluid, enthesis‐adjacent tissue and sacroiliac joint biopsies, with matched peripheral blood as a comparator. Pair AS cases with disease controls and healthy donors to define effect sizes.
Single‐Cell Multi‐Omics to Define Exhaustion Resistance
7.2
Use single‐cell RNA sequencing combined with surface‐protein indexing by cellular indexing of transcriptomes and epitopes by sequencing (CITE‐seq) to co‐measure transcriptomes and checkpoint or co‐stimulatory proteins in the same cells [81]. This enables an operational definition of exhaustion resistance as PD‐1^high cytotoxic CD8^+^ T cells with preserved effector programmes and low TOX‐module scores. Multiome profiling that measures gene expression and chromatin accessibility in the same cell can link resilience to accessible regulatory elements at loci such as TOX, TCF7 and STAT5 targets [82, 83]. Recommended outputs include gene‐module scores, chromatin peak enrichment, and joint clustering of RNA and ATAC features [84].
Spatial Context of Sustaining Signals
7.3
Apply spatial transcriptomics on sacroiliac or enthesis sections to localise IL‐15, B7 ligands and antigen‐presenting niches relative to PD‐1^high CD8^+^ T cell aggregates [85]. Report spatial co‐enrichment statistics and neighbourhood analyses to identify microenvironments that could maintain effector competence [86].
TCR Tracking and Evidence for Intermittent Stimulation
7.4
Perform 5′ single‐cell RNA sequencing (scRNA‐seq) with paired V(D)J to recover TCR sequences, quantify clonal expansion across blood and lesion and assess state transitions of dominant clonotypes. Use established algorithms such as GLIPH2 to infer shared antigen specificity groups [87]. A key prediction is that expanded lesion‐resident clones cycle through activation states without entering a stable TOX^high programme under in vivo conditions that are intermittent in antigen exposure.
Functional Metabolic Profiling
7.5
Quantify bioenergetics in sorted PD‐1^high CD8^+^ cells using extracellular flux assays to derive oxygen consumption and extracellular acidification parameters, ATP production rates from oxidative phosphorylation and glycolysis, and spare respiratory capacity [88]. Complement with SCENITH flow‐based profiling to map pathway dependencies at single‐cell resolution in fresh samples [89]. Report whether exhaustion‐resistant cells show preserved oxidative capacity and translation under pathway blockade.
Ex Vivo Perturbations That Test Each Axis
7.6
To interrogate the intermittent TCR axis, compare pulsatile anti‐CD3 stimulation (short pulses separated by rest intervals) with continuous stimulation and quantify cytotoxic readouts, induction of TOX and NR4A modules, and state shifts of dominant clonotypes. For the metabolic axis, titrate inhibitors of mTOR or mitochondrial respiration and attempt rescue with agents that enhance mitochondrial biogenesis; read out effector function together with oxygen‐consumption rate (OCR), extracellular‐acidification rate (ECAR), ATP production and spare respiratory capacity. For the IL‐15/CD28 axis, block IL‐15Rα or Janus kinase (JAK)‐STAT signalling or inhibit CD28–B7 co‐stimulation and determine whether effector competence declines with a concomitant rise in TOX‐programme scores [90]. These perturbations nominate pharmacodynamic readouts (e.g., pSTAT5 in CD8^+^ T cells, OCR/ECAR and TOX‐module scores) that can be translated into early‐phase trials.
Falsification Criteria and Biomarkers
7.7
The model would be falsified if PD‐1^high cytotoxic CD8^+^ T cells in lesions uniformly display TOX^high programmes with low mitochondrial fitness when measured by single‐cell multi‐omics and functional assays or if targeted blockade of IL‐15/JAK or CD28–B7 signals fails to reduce effector competence ex vivo. Positive validation would nominate biomarkers such as pSTAT5 levels, IL‐15 spatial density near PD‐1^high clones, high spare respiratory capacity and accessibility at enhancer elements near TCF7 and cytotoxic loci for future patient stratification.
Applicability to AS and Clinical Implications
8
Ankylosing spondylitis presents a distinctive immunologic milieu that aligns with the proposed three‐axis model. HLA‐B27 biology and the enthesis organ architecture create niches for local immune activation; human enthesis contains resident lymphocytes that respond to interleukin‐23 and γδ T cells capable of interleukin‐17 production, supporting site‐restricted stimulation [14, 15, 16]. A gut–joint axis is also evident: subclinical intestinal inflammation is frequent and can be tracked by faecal calprotectin, and an AS‐associated ileal microbial signature has been described [17, 18, 19]. Repetitive biomechanical stress at entheses provides episodic danger cues and antigen release, favouring intermittent and tissue‐confined TCR engagement rather than uniform, chronic exposure [20].
Understanding how CD8^+^ T cells resist exhaustion suggests testable therapeutic strategies in AS. Janus kinase inhibitors intercept signalling from common γ‐chain cytokines, including interleukin‐15, which can provide survival and metabolic support to cytotoxic T cells; consistent with this mechanism, JAK inhibition has shown clinical benefit in axial spondyloarthritis [90]. Candidate pharmacodynamic markers derived from our roadmap include pSTAT5 in lesion‐derived CD8^+^ T cells, IL‐15 spatial density near PD‐1^high aggregates and spare respiratory capacity. In phase 3 studies, tofacitinib improved signs and symptoms of active ankylosing spondylitis versus placebo, and upadacitinib achieved rapid responses with sustained efficacy across radiographic and non‐radiographic cohorts [91, 92, 93]. These data support targeting cytokine networks that sustain exhaustion‐resistant CD8^+^ T cell pools and motivate hypothesis‐driven trials of agents that modulate the interleukin‐15–JAK–STAT5 axis or CD28–B7 co‐stimulation in carefully selected patients. Safety considerations reported with JAK inhibition in other immune diseases should be prospectively monitored in AS [94].
Illustrative examples for JAK inhibition include upadacitinib in radiographic and non‐radiographic axSpA: SELECT‐AXIS 1 (phase 2/3; long‐term extension to 2 years; NCT03178487) [95] and SELECT‐AXIS 2 (phase 3; NCT04169373) [92], as well as tofacitinib (phase 3; NCT03502616) [91]. In addition, filgotinib has progressed to a phase 3 axSpA programme (OLINGUITO; e.g., NCT05785611; EU CTIS 2022–501 354–10‐01) with positive topline results reported in July 2025. For IL‐15 antagonism, active development is underway in related immune conditions, including ordesekimab/PRV‐015 in non‐responsive coeliac disease (NCT04424927; prior AMG‐714 study NCT02637141) and CALY‐002 in coeliac disease/eosinophilic oesophagitis (NCT04593251) [96, 97]. To our knowledge, no interventional trials of IL‐15 antagonists in axial spondyloarthritis were registered at the time of writing.
Future Directions
9
Validating the Core Molecular Mechanisms
9.1
First, it is crucial to validate the core molecular mechanisms proposed in our model. This will involve using advanced techniques like spatial transcriptomics to map the niches where these cells reside and identify their sustaining signals [85]. Furthermore, detailed epigenetic analyses, such as assays for transposase‐accessible chromatin using sequencing (ATAC‐seq) on sorted PD‐1^+^TOX^−^ cells, are needed to define their unique chromatin landscape and reveal novel regulatory targets. Finally, the causative roles of the IL‐15/CD28 axis and metabolic pathways must be confirmed using AS‐relevant preclinical models through genetic or pharmacological interventions.
Translational Research and Biomarker Development
9.2
A key goal is to determine whether the frequency or functional signature of circulating or synovial exhaustion‐resistant CD8^+^ T cells can serve as biomarkers for disease activity, structural progression or treatment response. Prospective, biomarker‐enriched studies should link target engagement to clinical outcomes. Candidate readouts include pSTAT5 in lesion‐derived CD8^+^ T cells, spatial density of interleukin‐15–producing niches near PD‐1^high CD8^+^ aggregates, spare respiratory capacity and ATP production rates from extracellular flux analysis, and transcriptomic module scores such as TOX‐ and TCF7‐associated programmes with paired TCR tracking. Monitoring how this compartment changes under approved and experimental therapies will clarify mechanisms of action and resistance.
Developing Novel Therapeutic Strategies
9.3
This framework also inspires the development of novel therapeutic strategies. Beyond just blocking inflammatory outputs, future approaches could aim to reprogram the pathogenic T cells themselves. For instance, exploring ways to target the supporting axes, such as with IL‐15 receptor antagonists, represents a direct application of our model. Alternatively, strategies aimed at therapeutically inducing exhaustion, perhaps by modulating the expression or activity of key transcription factors like TOX [28], could offer a way to selectively silence these pathogenic cells without broad immunosuppression.
Outlook
10
The conceptual framework of ‘CD8^+^ T‐cell exhaustion‐resistant’ provides a novel lens through which to understand the persistent immune activation and structural progression in ankylosing spondylitis. This theory brings CD8^+^ T cells to the centre of AS immunopathology, complementing the classical Th17/IL‐17 axis. While previous models often emphasised HLA‐B27 misfolding or innate immune activation, our review repositions HLA‐B27 as a sustained antigen‐presenting molecule that expands exhaustion‐resistant CD8^+^ clones, thereby fuelling adaptive immune dysregulation. The documented flares of arthritis in AS patients undergoing checkpoint inhibitor therapy underscore the clinical relevance of this mechanism.
In therapeutic terms, the concept of exhaustion‐resistant encourages a shift from rejuvenating immunity—as in cancer—to promoting controlled attrition of autoreactive CD8^+^ T cells. Strategies such as driving TOX/NR4A expression, locally enhancing PD‐1 signalling or disrupting IL‐15–mediated metabolic support hold promise for selectively silencing these pathogenic T cells without compromising systemic defence. JAK inhibitors, already efficacious in AS, may partly exert their effects by attenuating the very cytokine circuits that sustain exhaustion‐resistant.
Still, this model remains theoretical and demands further empirical support. Future studies should dissect the epigenetic and metabolic signatures of exhaustion‐resistant CD8^+^ T cells, clarify their interaction with the osteoimmune microenvironment, and evaluate targeted interventions in animal models. Importantly, the spatial organisation and niche‐specific signals that foster this resistant state warrant close investigation.
In summary, exhaustion‐resistant CD8^+^ T cells may constitute a missing link between persistent inflammation, structural damage and immunotherapy refractoriness in AS. Anchored in both molecular insight and clinical observation, this framework may not only refine disease taxonomy but may also help guide the next generation of immune‐modulating therapies in spondyloarthritides.
Author Contributions
Xuhong Zhang: conceived the structure of the review and conducted the literature search. Lu Jia and Xueni Lin: drafted the initial manuscript and prepared the figures. Lamei Zhou: contributed to the critical revision of the manuscript. All authors read and approved the final version of the manuscript.
Disclosure
The authors have nothing to report.
Ethics Statement
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1J. D. Taurog , A. Chhabra , and R. A. Colbert , “Ankylosing Spondylitis and Axial Spondyloarthritis,” New England Journal of Medicine 374 (2016): 2563–2574.27355535 10.1056/NEJ Mra 1406182 · doi ↗ · pubmed ↗
- 2M. A. Brown , T. Kenna , and B. P. Wordsworth , “Genetics of Ankylosing Spondylitis—Insights Into Pathogenesis,” Nature Reviews Rheumatology 12 (2016): 81–91.26439405 10.1038/nrrheum.2015.133 · doi ↗ · pubmed ↗
- 3P. Atagunduz , H. Appel , W. Kuon , et al., “HLA–B 27–Restricted CD 8+ T Cell Response to Cartilage‐Derived Self Peptides in Ankylosing Spondylitis,” Arthritis and Rheumatism 52 (2005): 892–901, 10.1002/art.20948.15751060 · doi ↗ · pubmed ↗
- 4R. A. Colbert , T. M. Tran , and G. Layh‐Schmitt , “HLA‐B 27 Misfolding and Ankylosing Spondylitis,” Molecular Immunology 57 (2014): 44–51.23993278 10.1016/j.molimm.2013.07.013PMC 3857088 · doi ↗ · pubmed ↗
- 5I. Wong‐Baeza , A. Ridley , J. Shaw , et al., “KIR 3DL 2 Binds to HLA‐B 27 Dimers and Free H Chains More Strongly Than Other HLA Class I and Promotes the Expansion of T Cells in Ankylosing Spondylitis,” Journal of Immunology 190 (2013): 3216–3224.10.4049/jimmunol.1202926 PMC 373609423440420 · doi ↗ · pubmed ↗
- 6K. Pavelka , A. J. Kivitz , E. Dokoupilova , et al., “Secukinumab 150/300 Mg Provides Sustained Improvements in the Signs and Symptoms of Active Ankylosing Spondylitis: 3‐Year Results From the Phase 3 measure 3 Study,” ACR Open Rheumatology 2 (2020): 119–127.31957970 10.1002/acr 2.11102 PMC 7011421 · doi ↗ · pubmed ↗
- 7R. Micheroli , S. Kissling , K. Bürki , et al., “Sacroiliac Joint Radiographic Progression in Axial Spondyloarthritis Is Retarded by the Therapeutic Use of TNF Inhibitors: 12‐Year Data From the SCQM Registry,” RMD Open 8 (2022): e 002551.36270744 10.1136/rmdopen-2022-002551 PMC 9594572 · doi ↗ · pubmed ↗
- 8M. Tang , Z. Qaiyum , M. Lim , and R. D. Inman , “Single Cell Immune Profiling in Ankylosing Spondylitis Reveals Resistance of CD 8+ T Cells to Immune Exhaustion,” Iscience 28 (2025): 112715.40687839 10.1016/j.isci.2025.112715 PMC 12272782 · doi ↗ · pubmed ↗