Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9): The Multifaceted Biology, Diseases, and Pharmaceutical Interventions
Jia Kuang, Lei Hao, Meibiao Zhang, Zhao Yang

TL;DR
PCSK9 inhibitors lower cholesterol and may help prevent and treat ischemic stroke by multiple mechanisms, offering new hope for stroke therapy.
Contribution
This review systematically consolidates the mechanisms and clinical potential of PCSK9 inhibitors in stroke prevention and treatment.
Findings
PCSK9 inhibitors reduce LDL cholesterol by blocking PCSK9's degradation of LDL receptors.
They offer neuroprotective and vasculoprotective effects through LDLR-independent pathways.
PCSK9 inhibitors may improve endothelial function and modulate immune and metabolic responses.
Abstract
Ischemic stroke remains a leading cause of global disability and death. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have emerged as potent lipid‐lowering agents with expanding therapeutic potential. Beyond robust low‐density lipoprotein cholesterol reduction, accumulating evidence suggests these drugs may confer benefits in ischemic stroke prevention and management. However, challenges regarding accessibility, real‐world efficacy, and integration into combination therapies persist, necessitating a comprehensive evidence synthesis. This review systematically consolidates the molecular mechanisms of PCSK9 inhibition and classifies current inhibitors. We delineate recent preclinical advances underscoring their neuroprotective and vasculoprotective effects, alongside critical findings from major clinical trials. These developments highlight promising avenues for both…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
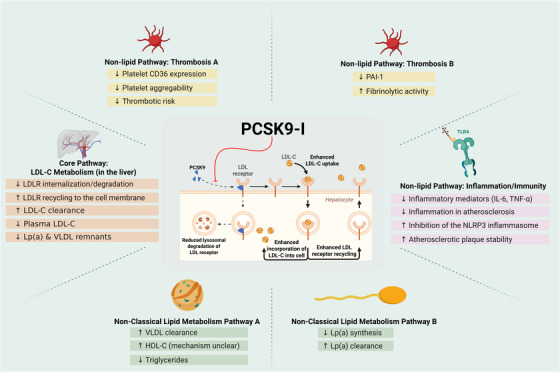 FIGURE 1
FIGURE 1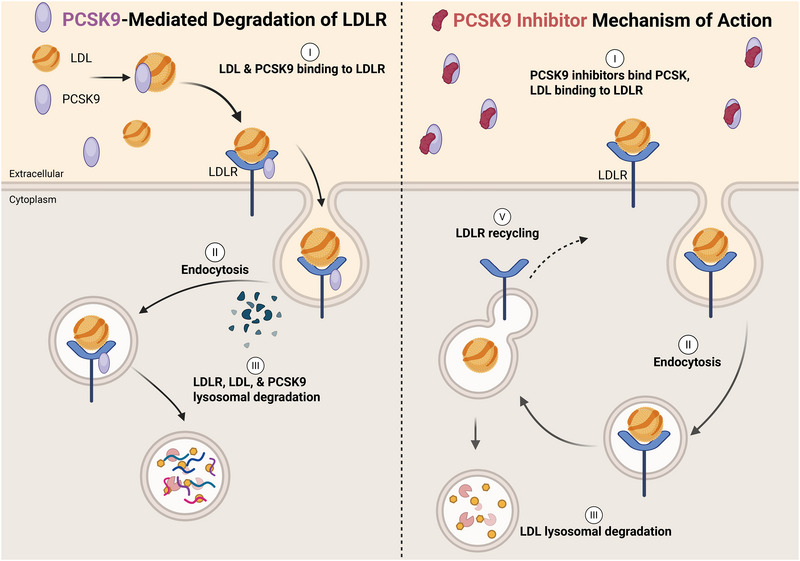 FIGURE 2
FIGURE 2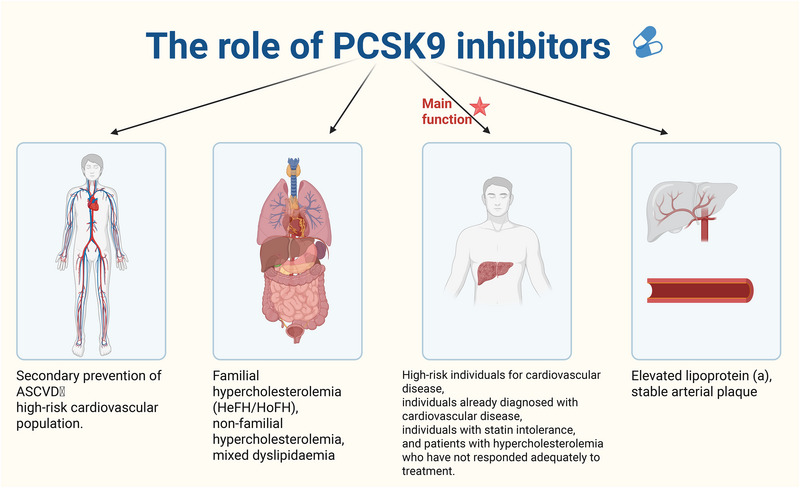 FIGURE 3
FIGURE 3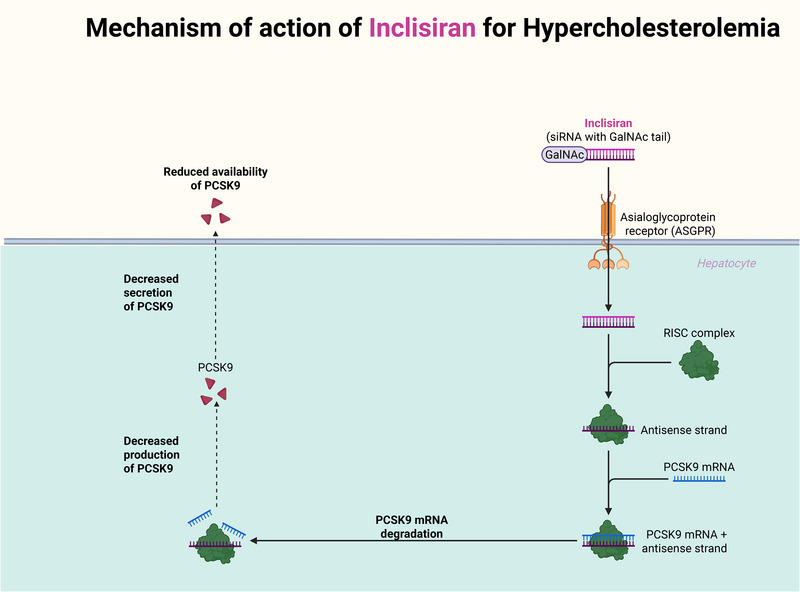 FIGURE 4
FIGURE 4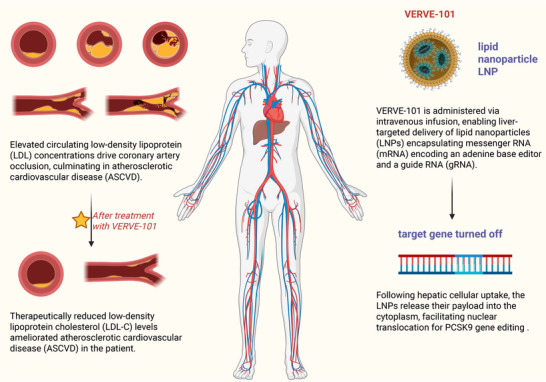 FIGURE 5
FIGURE 5| Drug name | NCT/ID | Indications | Global top stage |
|---|---|---|---|
| Ongericimab | 05325203 |
Approved (3): primary hypercholesterolemia; dyslipidemia; hyperlipidemia Investigational (7): heterozygous familial hypercholesterolemia; mixed hyperlipidemia; pure familial hypercholesterolemia; nonfamilial hypercholesterolemia Clinical stage III: hypercholesterolemia Clinical stage II: cardiovascular disease Clinical application: tumors | Approved for marketing (2024‐10‐09) |
| Undisclosed, AFFiRiS AG (PCSK9, hypercholesterolemia, biepitopic) | 02508896 | In progress: (preclinical) hypercholesterolemia | Preclinical (2024‐12‐13) |
| SAL‐003 | None |
In research (4): Clinical stage III: mixed hyperlipidemia; hypercholesterolemia Clinical stage I: hyperlipidemia Clinical application: dyslipidemia | Clinical phase III (2023‐07‐13) |
| Evolocumab | 02304484 |
In progress (11): Registry application: cardiovascular events Clinical phase III: type 2 diabetes mellitus; HIV infection; ST‐segment elevation myocardial infarction; coronary artery disease; arteritis; acute coronary syndrome; heterozygous familial hypercholesterolemia; hyperlipidemia Clinical stage II: metastatic desmoplasia‐sensitive prostate cancer Clinical phase I: myocardial infarction Approved (10): atherosclerosis; atherosclerotic cardiovascular disease; primary hypercholesterolemia; familial hypercholesterolemia; heart disease; cardiovascular disease; mixed hyperlipidemia; heterozygous familial hypercholesterolemia; dyslipidemia; hypercholesterolemia | Approval to market (2015‐07‐17) |
| B‐1655 | 02613871 | In research (2): clinical phase I: hypercholesterolemia; hyperlipidemia | Clinical phase I (2021‐01‐26) |
| Recombinant human anti‐PCSK9 monoclonal antibody | 2000031373 | In progress: (clinical phase I) hypercholesterolemia | Clinical phase I (2020‐05‐06) |
| ANTI PCSK9 MAB | 03355027 | In progress: (clinical phase I) hypercholesterolemia | Clinical phase I (2018‐04‐13) |
| DS‐9001 | None | Termination: dyslipidemia | Clinical phase I (2017‐05‐11) |
| Undisclosed, Aanastra Inc (PCSK9, familial hypercholestrolemia, peptide–mRNA) | None | In progress: (preclinical) hypercholesterolemia | Preclinical |
| PBGENE–PCSK9 | None | In progress: (preclinical) familial hypercholesterolemia | Preclinical |
| SGPCSK9i | None | In progress: (preclinical) hypercholesterolemia | Preclinical |
| Trial | NTC | Study Drug | Number of PCSK treated | Number of controls | Age | DM (%) | HT (%) | Smoking (%) | Number of strokes | |
|---|---|---|---|---|---|---|---|---|---|---|
| Treated | Controls | |||||||||
| OSLER I‐II | 01439880 | Evolocumab | 2976 | 1489 | 58.1 | 13.0 | 52 | 15.5 | 1 | 3 |
| ODYSSEY long term | 01507831 | Alirocumab | 1553 | 788 | 60.4 | 34.5 | − | 20.5 | 9 | 2 |
| Glagov | 01813422 | Evolocumab | 484 | 484 | 59.8 | 20.5 | 83 | 23.3 | 2 | 3 |
| FOURIER | 01764633 | Evolocumab | 13, 784 | 13, 780 | 62.5 | 36.5 | 80.0 | 28.3 | 207 | 262 |
| SPIRE I‐II | 01968980 | Bococizumab | 13, 720 | 13, 718 | 63.3 | 47.5 | 80.8 | 24.5 | 45 | 75 |
| ODYSSEY | 01954394 | Alirocumab | 9462 | 9462 | 58.5 | 29.0 | 65.0 | 24.0 | 111 | 152 |
| Total: | − | − | 41, 979 | 39, 721 | 60.5 | 32.1 | 69.5 | 22.6 | 375 | 497 |
- —Chongqing Science and Health Joint Medical Research Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLipoproteins and Cardiovascular Health · Acute Ischemic Stroke Management · Antiplatelet Therapy and Cardiovascular Diseases
Introduction
1
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a pivotal serine protease primarily synthesized in the liver, playing a critical role in cholesterol metabolism by promoting the degradation of hepatic low‐density lipoprotein receptors (LDLRs) [1, 2]. Structurally, PCSK9 consists of a signal peptide, a prodomain, a catalytic subunit, and a C‐terminal domain, with its function tightly regulated by autocatalytic processing. Since its initial discovery in 2003 through genetic linkage analyses, PCSK9 has rapidly emerged as a central player in lipid homeostasis. Landmark studies, including the identification of gain‐of‐function mutations associated with familial hypercholesterolemia (FH) and loss‐of‐function (LOF) variants linked to hypocholesterolemia and reduced cardiovascular risk, established its therapeutic significance, ultimately culminating in the development of monoclonal antibody inhibitors.
Beyond its canonical role in LDL cholesterol regulation, accumulating evidence underscores the involvement of PCSK9 in extrahepatic pathological processes—particularly in cerebrovascular and neurovascular diseases. PCSK9 is expressed in vascular smooth muscle cells (VSMCs) and endothelial cells, where it exacerbates inflammatory responses, promotes atherosclerotic plaque progression and instability, and enhances platelet activation and thrombogenesis. These pleiotropic mechanisms position PCSK9 at the intersection of dyslipidemia, inflammation, and thrombosis—key drivers of ischemic stroke, which remains a leading cause of mortality and long‐term disability worldwide [3, 4, 5].
Despite the established efficacy of statins, significant residual cardiovascular risk persists, underscoring the need for novel therapeutic strategies. PCSK9 inhibitors (PCSK9‐i), which achieve profound and sustained LDL cholesterol (LDL‐C) reduction, have demonstrated promising pleiotropic benefits including plaque stabilization, endothelial protection, and antithrombotic effects [6]. Nevertheless, current clinical management has yet to fully embrace a mechanism‐based, precision medicine approach for PCSK9 modulation. Important challenges—such as treatment accessibility, real‐world effectiveness, optimal timing in acute settings, and integration with combination therapies—remain unresolved [7, 8, 9, 10].
This review seeks to synthesize contemporary evidence on the therapeutic potential of PCSK9 inhibition in ischemic stroke, spanning from molecular mechanisms to clinical applications. We begin by delineating the biochemical and structural features of PCSK9 and summarizing milestone discoveries in its research trajectory. We then systematically classify existing and emerging PCSK9‐targeting agents, including monoclonal antibodies, oral inhibitors, and epigenetic editing therapies. Furthermore, we consolidate preclinical and clinical findings supporting their roles in neuroprotection, vasculoprotection, and improved functional outcomes after stroke. Finally, we discuss future directions involving personalized treatment strategies and combinatorial regimens, aiming to pave the way for a new era of precision medicine in neurovascular care.
PCSK9 Biology and Physiology
2
PCSK9, a hepatocyte‐derived serine protease, regulates LDL‐C by binding the LDL receptor (LDLR), redirecting it to lysosomal degradation instead of recycling, thereby reducing hepatic LDL‐C clearance. Gain‐of‐function mutations elevate cardiovascular risk, while LOF variants lower LDL‐C and atherosclerosis susceptibility. Mechanistically, PCSK9 disrupts sorting nexin 17 (SNX17)‐mediated LDLR recycling in endosomes, and mutations impairing this pathway confer resistance to PCSK9‐i. Beyond lipid metabolism, PCSK9 promotes inflammation (e.g., NLRP3 activation via lipid accumulation), thrombosis (e.g., platelet activation via CD36), and plaque vulnerability. PCSK9‐i (e.g., evolocumab, alirocumab) neutralize circulating PCSK9, restoring LDLR recycling and reducing LDL‐C by 50–60%, while pleiotropic effects (e.g., plaque stabilization, anti‐inflammation) contribute to cardiovascular risk reduction independently of LDL‐C lowering (Figure 1).
Summary of PCSK9 inhibitor mechanisms. PCSK9 inhibitors (PCSK9‐i) primarily lower LDL‐cholesterol (LDL‐C) by blocking PCSK9‐mediated degradation of hepatic LDL receptors (LDLR), enhancing LDL clearance. Beyond LDLR‐dependent effects, PCSK9‐i exert multiple benefits via LDLR‐independent pathways. These include anti‐inflammatory effects (suppressing TLR4/NF‐κB, NLRP3 inflammasome, and cytokine release), antioxidant actions (reducing NOX‐derived ROS and enhancing Nrf2/HO‐1), and improved endothelial function (via eNOS/NO upregulation and reduced adhesion molecules). Additionally, PCSK9‐i may modulate immune responses (reducing macrophage CD36 and T‐cell activation), thrombosis (lowering platelet activity and tissue factor), and metabolic pathways (potentially improving insulin sensitivity via SIRT1/AMPK). They also influence plaque stability by decreasing smooth muscle cell proliferation and oxidative stress.
Biosynthesis and Structural Basis of Function
2.1
PCSK9, a hepatocyte‐derived serine protease belonging to the proprotein convertase family, plays a pivotal role in modulating protein homeostasis through proteolytic processing and degradation of secreted proteins [11, 12]. Its primary biological function centers on regulating LDL cholesterol (LDL‐C) metabolism via interaction with the LDLR (Figure 2). Mechanistically, PCSK9 binds to LDLR on hepatocyte membranes, redirecting the receptor toward lysosomal degradation rather than cellular recycling, thereby limiting hepatic LDL‐C clearance [13, 14]. This intervention reduces hepatic LDLR density by 50–70%, diminishes LDL‐C clearance capacity by 30–40%, and elevates circulating LDL‐C levels [15, 16, 17, 18].
Primary biological function of PCSK9 inhibitors. PCSK9 inhibitors (PCSK9‐i) primarily function by regulating LDL cholesterol (LDL‐C) metabolism through their interaction with the LDL receptor (LDLR). By binding to and inhibiting circulating PCSK9, these therapeutics prevent PCSK9‐mediated degradation of LDLR in hepatocytes. This stabilization of LDLR enhances hepatic clearance of LDL‐C from plasma, leading to significant reductions in circulating LDL‐C levels. Beyond this central mechanism, PCSK9‐i exhibit secondary effects on lipid metabolism, including modest reductions in lipoprotein(a) [Lp(a)] and very‐low‐density lipoprotein (VLDL) remnants. While their primary action is LDLR dependent, emerging evidence suggests potential LDLR‐independent effects on inflammatory pathways, endothelial function, and platelet activity.
Gain‐of‐function mutations (e.g., D374Y, S127R) amplify PCSK9 activity, causing autosomal dominant hypercholesterolemia (LDL‐C > 190 mg/dL) and accelerating atherosclerosis progression. Conversely, LOF variants (e.g., R46L, Y142X) reduce circulating PCSK9 by 40%, lower LDL‐C by 15–28%, and decrease cardiovascular risk by 47% [19, 20, 21]. These findings not only validate PCSK9 as a critical therapeutic target but also highlight its potential for personalized interventions in patients with heightened sensitivity to LDL‐C atherogenicity [22]. PCSK9‐i constitute a significant therapeutic advancement in lipid management, primarily targeting the LDLR degradation pathway [23]. Consequently, managing these patients necessitates genetic diagnosis independent of LDLR function assessment and consideration of alternative therapeutic agents. By neutralizing circulating PCSK9 via monoclonal antibody administration (e.g., evolocumab, alirocumab), PCSK9‐i disrupt the PCSK9–LDLR interaction, thereby preserving surface LDLR density on hepatocyte membranes. This augmentation of LDLR availability significantly increases the hepatic clearance rate of LDL particles, resulting in robust plasma LDL‐C reduction of 50–60%. This efficacy profile has been conclusively demonstrated in pivotal cardiovascular outcomes trials, including FOURIER (in patients with atherosclerotic cardiovascular disease [ASCVD]) and ODYSSEY OUTCOMES (in high‐risk cardiovascular cohorts) [24, 25, 26, 27, 28].
It is widely held within the lipidology community that the primary cardiovascular benefits of PCSK9 inhibition derive significantly from robust reductions in LDL‐C. Proteomic analyses, exemplified by the Systematic Protein Investigative Research Environment (SPIRE) trial, demonstrate that the modulatory effects of PCSK9‐i on circulating inflammatory markers are wholly concordant with the magnitude of achieved LDL‐C lowering. Crucially, no distinct inflammatory signaling pathways independent of LDL‐C reduction have been identified. Genetic studies of naturally occurring PCSK9 LOF variants provide robust corroborative evidence, strongly supporting LDL‐C reduction as the principal mediator of cardiovascular protection.
However, while the canonical mechanism of action (MoA) of PCSK9‐i involves enhanced recycling and stabilization of the LDLR, emerging mechanistic investigations suggest potential contributions from additional, noncanonical pathways that may operate independently of LDL‐C reduction. From a clinical perspective, the paramount therapeutic objective remains the attainment of guideline‐recommended LDL‐C targets. Any potential ancillary benefits arising from mechanistic nuances are likely to constitute secondary considerations, potentially offering context‐specific advantages beyond LDL‐C lowering per se [29].
Canonical Role in LDLR Recycling and Cholesterol Homeostasis
2.2
Compartmentalized LDLR Trafficking: SNX17‐Dependent Recycling Pathway
2.2.1
Research elucidating the SNX17‐dependent LDLR recycling pathway has established that PCSK9 binding prevents LDLR acidification‐induced conformational changes within endosomal compartments. This impairment disrupts critical binding interactions between SNX17 and the evolutionarily conserved “NPxY” endocytic sorting motif in the LDLR cytosolic domain. Consequently, LDLR trafficking to recycling endosomes is compromised, resulting in receptor retention within endolysosomal compartments and proteolytic degradation by lysosome‐resident acid hydrolases.
Notably, FH patients harboring LDLR LOF variants impacting intracellular trafficking pathways exhibit diminished therapeutic responsiveness to PCSK9‐i. This pharmacoresistance necessitates comprehensive genetic characterization independent of standard LDLR functional assays and mandates consideration of alternative lipid‐lowering strategies. These clinical observations substantiate the fundamental role of compartmentalized LDLR trafficking regulation in cholesterol homeostasis.
Crucially, experimental SNX17 ablation in macrophages completely abrogates PCSK9‐mediated LDLR degradation, demonstrating that localized receptor recycling operates independently of systemic LDL‐cholesterol (LDL‐C) clearance pathways. This mechanistic distinction provides the molecular foundation whereby PCSK9 inhibition enhances macrophage uptake and degradation of oxidized LDL (oxLDL) particles through LDLR pathway modulation—even when plasma LDL‐C levels remain unaltered. The pathway demonstrates tissue‐specific regulation of lipoprotein metabolism beyond hepatic clearance mechanisms [30, 31].
Plaque‐Specific Macrophage Reprogramming: LDLR‐Independent Modulation of Atherogenic Mechanisms
2.2.2
Transplantation of bone marrow overexpressing human PCSK9 (hPCSK9) into apolipoprotein E‐deficient (apoE^−^/^−^) mice—which retain functional LDLR expression—demonstrated specific hPCSK9 accumulation within atherosclerotic plaques. This localized overexpression induced significant downregulation of both LDLR and LDLR‐related protein 1 (LRP1) on macrophage surfaces, markedly compromising oxLDL clearance capacity [32, 33]. Administration of PCSK9‐i reversed this phenotype, restoring macrophage phagocytic function and reducing foam cell formation. Notably, parallel experiments in LDLR‐deficient (LDLR^−^/^−^) recipients revealed that circulating PCSK9 propagates macrophage activation and accelerates venous graft lesion progression through LDLR‐independent pathways.
These observations indicate that targeted LDLR upregulation within plaque‐resident macrophages improves intraplaque lipid homeostasis independently of systemic lipid parameters. Critically, PCSK9‐i achieve three synergistic antiatherogenic effects at the plaque level: specific upregulation of macrophage LDLR expression; enhanced oxLDL efflux and catabolism; attenuation of NLRP3 inflammasome activation. Importantly, these localized beneficial effects persist even in contexts of suboptimal systemic LDL‐C concentrations [34, 35]. This suggests that modulating macrophage LDLR expression within atherosclerotic lesions confers therapeutic advantages without necessitating further reduction of circulating LDL‐C levels, highlighting a distinct dimension of PCSK9 inhibition beyond conventional lipid management.
Anti‐Inflammatory Effects Beyond LDL‐C Reduction: Pleiotropic Plaque Stabilization
2.2.3
In murine models with systemically controlled LDL‐C concentrations, local PCSK9 inhibitor administration decreased inflammatory monocyte infiltration into atherosclerotic plaques by 32% while simultaneously enhancing fibrous cap stability and collagen content [36]. These findings demonstrate that the anti‐inflammatory and plaque‐stabilizing properties of PCSK9 inhibition operate independently of circulating lipoprotein levels, establishing a molecular foundation for their pleiotropic vasculoprotective actions beyond conventional lipid‐lowering benefits. Concurrently, Gynostemma pentaphyllum‐derived total saponins modulate atherosclerotic pathophysiology through: suppression of hepatic PCSK9 transcription; activation of macrophage autophagy‐lysosomal degradation pathways (lipophagy); enhanced LDLR recycling via SNX17 upregulation; reduction of intracellular cholesteryl ester accumulation; inhibition of NLRP3 inflammasome‐mediated cytokine secretion [37, 38, 39, 40].
Collectively, these mechanistic insights substantiate the non‐lipid‐lowering therapeutic benefits of PCSK9‐i—particularly in patients achieving very low LDL‐C levels (<55 mg/dL). This has distinct clinical relevance for: FH patients exhibiting pharmacoresistance to conventional therapies; individuals with high‐risk plaques demonstrating active inflammatory signatures.
Noncanonical (Pleiotropic) Functions in Inflammation, Thrombosis, and Immunity
2.3
LDLR‐Dependent Inflammasome Priming via Intracellular Lipid Accumulation
2.3.1
PCSK9 significantly enhances mRNA expression of proinflammatory cytokines tumor necrosis factor (TNF)‐α and interleukin (IL)‐1β (increased by 40–45%) while concurrently elevating anti‐inflammatory mediators IL‐10 and Arg1 (30–44% increase) in lipopolysaccharide‐stimulated macrophages. This bidirectional immunomodulatory effect demonstrates strict LDLR dependence, as evidenced by complete abrogation in LDLR^−^/^−^ macrophages.
Through LDLR downregulation, PCSK9 impairs macrophage clearance of oxLDL, resulting in intracellular cholesterol crystallization. This lipid accumulation triggers NLRP3 inflammasome assembly and caspase‐1 activation, establishing a direct pathophysiological link between impaired cholesterol efflux and sterile inflammation.
Pharmacological PCSK9 inhibition attenuates this inflammatory cascade by restoring macrophage LDLR surface expression, thereby enhancing cholesterol efflux capacity and suppressing NLRP3 inflammasome activity independently of systemic lipid parameters [41, 42].
Pathophysiological Integration of PCSK9 in Atherosclerotic Plaques
2.3.2
PCSK9 demonstrates significant extrahepatic localization within arterial wall microenvironments, where it exerts pronounced proatherogenic influence. These local effects critically drive plaque vulnerability through distinct pathomorphological mechanisms that compromise structural integrity and compositional stability of advanced atheromata.
Beyond its canonical regulation of hepatic LDLR recycling and systemic lipid homeostasis, PCSK9 participates in diverse vascular pathophysiological cascades: Amplification of proinflammatory signaling cascades; modulation of VSMC phenotypic switching; promotion of VSMC proliferative and migratory phenotypes; enhancement of thrombogenicity through platelet reactivity and coagulation cascade modulation.
Proinflammatory Mechanisms of PCSK9 in Atherogenesis
2.3.3
Epidemiological analyses have revealed a significant positive correlation between circulating PCSK9 levels and systemic inflammatory biomarkers, including high‐sensitivity C‐reactive protein (hs‐CRP), underscoring its role in modulating inflammatory responses [11, 43, 44]. Importantly, clinical studies of PCSK9‐i demonstrate their capacity to remodel the inflammatory microenvironment within atherosclerotic plaques: immunohistochemical analyses show marked reductions in proinflammatory proteins (NLRP3, IL‐1β, TNF‐α) alongside increased collagen deposition, indicating enhanced plaque stability [45, 46]. The mechanistic link between PCSK9 activity and inflammatory biomarker elevation provides a compelling rationale for targeting this pathway in patients with high cardiovascular risk, particularly those exhibiting elevated hs‐CRP or unstable plaque morphology [45, 47]. Serial coronary computed tomography angiography studies demonstrate that combined administration of PCSK9‐i and statins attenuates total atheroma volume progression by 6–9% while enhancing fibrotic tissue content by 12–15%. These structural modifications correspond to a 27% reduction in recurrent major adverse cardiovascular events (MACEs) [48, 49, 50, 51].
Moreover, experimental studies in cholesteryl ester transfer protein‐expressing murine models reveal dramatic attenuation of plaque progression: 40–50% reduction in aortic arch lipid deposition; 35–45% decrease in systemic inflammatory biomarkers (IL‐6, TNF‐α); 25–30% increase in fibrous cap thickness; 40% reduction in necrotic core cross‐sectional area [52, 53].
Structural‐Driven VSMC Apoptosis: Necrotic Core Expansion via TLR4/Mitochondrial Axis
2.3.4
Structural studies demonstrate that the C‐terminal cysteine‐rich domain of PCSK9 binds to TLR4, activating the downstream NF‐κB signaling pathway, which drives the secretion of proinflammatory cytokines (e.g., IL‐6, TNF‐α) and promotes aberrant VSMC migration [45]. These mechanisms enable PCSK9 to regulate VSMC differentiation, proliferation, and motility, thereby accelerating atherosclerotic plaque development [52, 54]. Notably, PCSK9 induces mitochondrial dysfunction and apoptosis in VSMCs through morphology‐dependent pathways, processes strongly associated with increased vulnerability of carotid plaques [55, 56]. Cohort studies demonstrate a proportional relationship between serum PCSK9 levels and necrotic core volume within plaques (r = 0.58, p < 0.001), suggesting a direct mechanistic link to plaque destabilization [57]. The landmark STAINLAS trial further substantiates this association, revealing that elevated PCSK9 levels independently contribute to carotid plaque formation (OR 2.3, 95% confidence interval [CI] 1.6–3.4), even after adjusting for traditional risk factors [58]. These pathomorphological features develop independently of arterial lumen stenosis, emphasizing the critical role of PCSK9 in driving plaque vulnerability through inflammation‐mediated mechanisms [59].
Endothelial Salvage Pathways: Sirtuin‐Mediated Metabolic Revitalization
2.3.5
Vascular endothelial injury initiates a cascade of pathophysiological events: platelet adhesion, blood–brain barrier (BBB) compromise, and vasospasm—collectively promoting thrombogenesis and cerebral ischemia [60]. Clinical evidence demonstrates that PCSK9‐i preserve endothelial integrity through multimodal mechanisms: coronary vasculature: restoration of endothelial nitric oxide synthase (eNOS) activity enhances flow‐mediated dilation (35% improvement versus baseline) [61]; diabetic vasculopathy: combination therapy with glucose‐lowering agents augments endothelial progenitor cell (EPC) functionality (28% increase in EPC activity versus monotherapy) in type 2 diabetes patients [62, 63]. The landmark FOURIER trial established evolocumab‐mediated reduction in ischemic stroke incidence, attributable to both intensive LDL‐C reduction and documented suppression of platelet aggregation [64, 65, 66].
Preclinical studies indicate PCSK9‐i mitigate mitochondrial oxidative stress and improve vascular function via Sirtuin pathway activation (particularly SIRT3) in endothelial cells, mechanistically involving: upregulation of mitochondrial superoxide dismutase; enhanced endothelial metabolic flexibility [36, 67, 68, 69]. Additionally, immunomodulatory actions—including inhibition of oxLDL‐induced dendritic cell maturation—have been experimentally demonstrated, though their clinical translatability requires further validation.
Prothrombotic Circuitry: CD36‐Mediated Platelet Priming and Coagulation Cascade
2.3.6
Thrombosis, defined as the formation of a blood clot (thrombus) at sites of vascular injury or endothelial disruption, represents a critical pathological process underlying ischemic heart disease, ischemic stroke, and venous thromboembolism (VTE) [70, 71]. Beyond its role in atherosclerosis, PCSK9 enhances platelet reactivity by upregulating lipid‐mediated signaling pathways, thereby increasing blood hypercoagulability and thrombosis susceptibility [72]. Specifically, PCSK9 induces tissue factor expression in monocytes and elevates coagulation factor VIII (FVIII) levels, directly activating the extrinsic coagulation cascade [73]. Furthermore, PCSK9 interacts with the platelet CD36 receptor, amplifying platelet activation through lipid raft reorganization and prothrombotic signaling [72, 74, 75]. This interaction is functionally significant, as demonstrated by studies showing that CD36 knockdown or PCSK9‐i (e.g., evolocumab) substantially reduce platelet aggregation and thrombus formation in vivo [76].
Therapeutic interventions using PCSK9‐i counteract these effects through dual mechanisms: (1) reducing LDL‐C to mitigate lipid‐driven platelet hyperreactivity, and (2) blocking PCSK9–CD36 interactions to suppress direct platelet activation [76]. Additionally, PCSK9‐i moderately lower lipoprotein(a) [Lp(a)] levels, further attenuating thrombotic risk through multiply mechanisms [77].
PCSK9‐i exert atheroprotective effects through multifaceted mechanisms: Beyond their primary action of significantly reducing circulating LDL‐C levels via antagonism of PCSK9‐mediated LDLR catabolism, these agents target local arterial PCSK9 activity. They suppress proinflammatory signaling within the vascular wall, thereby attenuating PCSK9's adverse contributions to plaque composition—including enhanced macrophage infiltration and compromised fibrous cap integrityand modulating plaque vulnerability indices. Crucially, emerging evidence indicates direct inhibition of VSMC proliferation and migration, alongside potential interference with prothrombotic pathways. Consequently, PCSK9‐i synergistically ameliorate lipid profiles, promote plaque stabilization, and retard atherosclerosis progression through both lipid‐dependent and lipid‐independent mechanisms. This multiphasic activity translates to reduced incidence of MACEs [78].
Therapeutic Targeting of PCSK9: From Mechanisms to Agents
3
PCSK9‐i comprise two primary classes: monoclonal antibodies (e.g., evolocumab, alirocumab) and small‐interfering RNA (siRNA) therapeutics (e.g., inclisiran). Both enhance hepatic LDLR recycling, reducing LDL‐C by 50–60% and mitigating cardiovascular risk via pleiotropic pathways. Clinical trials (e.g., FOURIER, FOURIER‐OLE) demonstrate that evolocumab sustains LDL‐C suppression (<20 mg/dL), lowering cardiovascular mortality by 15% and ischemic stroke risk by 21% over 8.4 years. Imaging studies (GLAGOV, HUYGENS) further link PCSK9 inhibition to atherosclerotic plaque stabilization and regression. Inclisiran's biannual dosing enables prolonged LDL‐C control but is less optimal for rapid reduction. Synergy with statins achieves additive LDL‐C lowering (60–70%) and reduces VTE risk by 34%. Emerging CRISPR‐based epigenetic editing promises durable LDL‐C management, while existing classes support aggressive LDL‐C targets (<55 mg/dL) in high‐risk ASCVD patients.
Pharmacological Modalities: Monoclonal Antibodies, siRNA, and Emerging Platforms
3.1
PCSK9‐i comprise two principal classes: monoclonal antibodies (e.g., evolocumab, alirocumab) and siRNA therapeutics (e.g., inclisiran) (Table 1) [79, 80]. Both facilitate hepatic LDLR recycling, leading to a median reduction of 50–60% in circulating LDL‐C levels [81, 82, 83]. Beyond lipid lowering, these agents mitigate cardiovascular risk through pleiotropic pathways, solidifying their role as foundational therapies in contemporary cardiovascular disease management (Figure 3) [84, 85].
The role of PCSK9 inhibitors. PCSK9 inhibitors (PCSK9‐i) are a revolutionary class of therapeutics that primarily function by significantly lowering low‐density lipoprotein cholesterol (LDL‐C) levels through inhibition of PCSK9‐mediated degradation of hepatic LDL receptors, resulting in enhanced LDL particle clearance from circulation. Beyond their potent lipid‐lowering effects (achieving 50–60% reductions in LDL‐C), these agents provide robust cardiovascular protection by slowing atherosclerotic plaque progression, stabilizing vulnerable plaques, and reducing the incidence of MACEs in high‐risk patients. Additionally, PCSK9‐i exhibit multiple vascular benefits, including endothelial stabilization through improved nitric oxide bioavailability, reduced expression of adhesion molecules (VCAM‐1/ICAM‐1), and decreased endothelial inflammation. They also demonstrate anti‐inflammatory properties by modulating the NLRP3 inflammasome and suppressing proinflammatory cytokines, as well as antioxidant effects via upregulation of Nrf2/HO‐1 pathways.
Cardiovascular outcome trials, such as FOURIER and its open‐label extension FOURIER‐OLE, demonstrated that evolocumab achieves sustained LDL‐C reductions (median <20 mg/dL), alongside a 15% reduction in cardiovascular mortality (HR 0.85) and a 21% lower risk of ischemic stroke (HR 0.79) over a median follow‐up of 8.4 years [86, 87, 88]. Imaging studies including GLAGOV and HUYGENS further indicated that evolocumab promotes atherosclerotic plaque stabilization and regression by increasing fibrous cap thickness and reducing lipid core volume [89, 90].
Inclisiran, an siRNA therapy, targets and degrades PCSK9 mRNA within hepatocytes, enabling biannual dosing and prolonged LDL‐C reduction [91, 92, 93]. While both therapeutic classes achieve substantial LDL‐lowering, monoclonal antibodies are often preferred in scenarios requiring rapid LDL‐C reduction, such as in high‐risk patients with established ASCVD [94, 95, 96].
When combined with statins, PCSK9‐i exhibit synergistic effects. For instance, alirocumab added to high‐intensity statin therapy reduces LDL‐C by an additional 60–70% compared with statin monotherapy [97, 98, 99]. Meta‐analyses of randomized trials also indicate that this combination confers a 34% reduction in the risk of VTE (RR 0.66, 95% CI 0.49–0.89) [100, 101, 102].
Emerging strategies, such as CRISPR‐based epigenetic editing of PCSK9, hold potential for durable LDL‐C management but remain under investigation [103]. Despite differing mechanisms—extracellular neutralization for monoclonal antibodies versus intracellular gene silencing for siRNA therapies—both classes effectively achieve significant LDL‐C reduction, supporting a treatment paradigm aiming for LDL‐C levels below 55 mg/dL in high‐risk ASCVD patients [104, 105].
MoA: Neutralizing PCSK9 and Restoring LDLR Function
3.2
PCSK9‐i effectively ameliorate dyslipidemia by selectively binding to PCSK9, thereby preventing its interaction with the LDLR. This inhibition increases LDLR availability on hepatocytes, enhancing LDL‐C clearance and significantly reducing circulating LDL‐C levels by 50–60%, as demonstrated in clinical trials [106, 107, 108, 109].
The landmark FOURIER trial showed that PCSK9‐i not only achieve profound LDL‐C reduction (median LDL‐C <25 mg/dL) but also confer robust cardiovascular benefits, including a 15% reduction in cardiovascular mortality and a 21% lower risk of ischemic stroke. These benefits were sustained over long‐term follow‐up [86, 87].
In high‐risk patients with recent acute coronary syndromes (ACSs) and baseline LDL‐C near 70 mg/dL despite optimized statin therapy, PCSK9 inhibition provides additional clinical benefit when Lp(a) concentrations exceed 30 mg/dL, reducing MACEs by 23% [110, 111].
Thus, PCSK9‐i have emerged as a pivotal therapeutic option for lipid management, particularly in patients who fail to achieve adequate LDL‐C lowering with maximally tolerated statins and ezetimibe [112, 113, 114, 115]. Beyond lipid‐lowering, these agents may also improve stroke prognosis through pleiotropic mechanisms, such as plaque stabilization, endothelial function restoration, and attenuation of thrombotic risk [84, 85].
Preclinical Evidence of PCSK9 Inhibition Beyond Lipid‐Lowering
4
Preclinical studies have been instrumental in elucidating the multifaceted mechanisms by which PCSK9 inhibition confers protection beyond lipid lowering. The evidence can be categorized into four primary, interlinked pathways: direct neuroprotection, stabilization of the vascular compartment, and systemic anti‐inflammatory and immunomodulatory effects.
Antiatherosclerotic and Vasoprotective Effects
4.1
Preclinical studies in APOE*3Leiden.CETP transgenic mice, a well‐established model of atherosclerosis, revealed that PCSK9 gene knockdown exerts potent anti‐inflammatory effects. Genetic inhibition of PCSK9 significantly reduced TLR4 and NF‐κB expression in microglia (p < 0.01), concurrently lowering intraplaque levels of proinflammatory mediators such as IL‐1β and MCP‐1 (p < 0.05). This attenuation of vascular inflammation was mediated through dual mechanisms: suppression of the TLR4/NF‐κB signaling pathway and reduced uptake of oxLDL by macrophages. The study further demonstrated that PCSK9 inhibition decreases oxidative stress markers and enhances plaque stability by increasing collagen content, underscoring its therapeutic potential for mitigating inflammation‐driven atherosclerotic progression [116].
In a mouse model of abdominal aortic aneurysm (AAA), genetic knockout (Pcsk9^−^/^−^) or pharmacological inhibition (PCSK9‐targeting antibodies/siRNA) significantly attenuated vascular inflammation and structural degeneration. PCSK9 deficiency reduced aortic macrophage infiltration by 60–70% (p < 0.01) and elastin degradation by 40–50%, while simultaneously suppressing matrix metalloproteinase‐9 (MMP‐9) expression and enhancing the SIRT1/NF‐κB pathway, which preserved vascular integrity. These findings provide a robust preclinical foundation for translating PCSK9‐targeted therapies into clinical strategies for AAA management, particularly in patients with inflammatory‐driven vascular remodeling [36, 117].
Compared with prior research, this study more clearly delineates the crucial role of PCSK9 in plaque stabilization and extracellular matrix remodeling. Importantly, activation of the SIRT1/NF‐κB pathway upon PCSK9 inhibition suggests epigenetic regulation of inflammation, offering a deeper mechanistic basis for PCSK9‐targeted therapies. However, current evidence remains limited to preclinical models, and clinical translation necessitates validation of these mechanisms in human vascular tissues. Furthermore, exploring synergies between PCSK9‐i and other anti‐inflammatory agents (e.g., IL‐1β inhibitors) could help maximize the benefits of multipathway combination treatments.
Neuroprotective and BBB Stabilizing Effects
4.2
In atherosclerosis‐prone mouse models, the combination of alirocumab (a PCSK9 monoclonal antibody) with statins demonstrated neuroprotective potential by stabilizing atherosclerotic plaques and mitigating vascular endothelial damage through inhibition of inflammatory cytokines such as IL‐6 and TNF‐α, thereby indirectly preserving neurological function [118]. Similarly, in FH models, PCSK9 monoclonal antibodies exhibited neuroprotective effects by modulating the immune microenvironment, reducing neuroinflammation, and enhancing blood–brain barrier integrity [119].
In a mouse model of Alzheimer's disease, the PCSK9 inhibitor evolocumab demonstrated neuroprotective effects by enhancing BBB integrity. Mechanistically, evolocumab reduced MMP‐9 secretion through inhibition of microglial activation, thereby attenuating amyloid‐β (Aβ) deposition‐induced BBB disruption. These findings highlight a novel therapeutic avenue for neurodegenerative disorders, extending PCSK9‐i’ benefits beyond lipid modulation to direct neurovascular protection [120].
In murine models, evolocumab significantly reduces plasma LDL‐C without affecting cerebral cholesterol levels—a contrast to statins like atorvastatin, which penetrate the BBB and suppress brain cholesterol synthesis [121]. In vitro BBB models reveal that PCSK9‐i maintain barrier function by enhancing LRP1‐mediated Aβ clearance, independent of central cholesterol metabolism [122]. oxLDL‐induced endothelial damage via LOX‐1 receptor activation is mitigated by PCSK9‐i through dual pathways: reduction of oxLDL levels inhibits endothelial apoptosis and BBB disruption and attenuation of aortic inflammation in AAA models by suppressing macrophage infiltration and MMP‐9 activity, concurrently reducing serum TNF‐α and IL‐6 (p < 0.01) while upregulating tight junction proteins (e.g., claudin‐5) to fortify BBB integrity [36].
Elevated homocysteine levels, associated with plaque instability, are counteracted by PCSK9‐i through slowing intraplaque neovascularization and inflammation, indirectly stabilizing BBB integrity, and lowering oxLDL levels and inhibiting microglial activation to reduce neuroinflammation and endothelial dysfunction. These interconnected mechanisms form a comprehensive BBB protective network, positioning PCSK9‐i as unique therapeutic agents capable of addressing both vascular and neurological aspects of cerebrovascular disease [123].
Further insights emerged from studies using hyperlipidemic mice subjected to ischemic stroke via middle cerebral artery occlusion (MCAO). This intervention reduced infarct volume by 35–40% and improved functional recovery, underscoring the therapeutic potential of PCSK9 inhibition in stroke management [124]. Complementary research in a transient MCAO model revealed that PCSK9 inhibitor treatment upregulated glycoprotein nonmetastatic melanoma protein B, a neuroprotective factor, resulting in a 30% reduction in infarct volume and enhanced neuronal survival. This suggests that PCSK9‐i may amplify endogenous neuroprotective pathways, offering dual benefits in ischemia–reperfusion injury [125].
PCSK9‐i provide neuroprotection mainly by stabilizing the BBB and modulating immune responses. These actions support emerging strategies focused on cerebrovascular integrity. For instance, similar to sigma‐1 receptor agonists such as DMT, PCSK9‐i help reduce neuroinflammation and maintain barrier function by stabilizing tight junction proteins and inhibiting release of proinflammatory cytokines. Key pathways involved include CX43–PARP1–NAD⁺ and PI3K/AKT, which are crucial for BBB maintenance under disease conditions. By mitigating IL‐6 and TNF‐α‐mediated inflammation, PCSK9‐i highlight potential immunometabolic therapies for stroke and neurodegeneration. However, their exact mechanisms in neuroinflammatory cascades need further study. Future research should investigate whether PCSK9 regulation directly influences astrocyte activation or endothelial permeability. Additionally, since monoclonal antibodies poorly cross the intact BBB, clinical applications may require combination with permeabilization techniques like focused ultrasound or nanoparticle delivery to improve efficacy.
Antithrombotic Effects
4.3
In a venous thrombosis model induced by inferior vena cava ligation, PCSK9‐deficient mice exhibited a 40–50% reduction in thrombus size compared with wild‐type controls, accompanied by significantly lower serum soluble P‐selectin (sP‐selectin) levels (↓35%, p < 0.01). This suggests PCSK9 modulates thrombogenesis by enhancing platelet α‐granule release and endothelial activation [126]. Further mechanistic studies revealed that PCSK9 regulates thrombus formation via interactions with platelet membrane receptors (e.g., CD36) and endothelial adhesion molecules (e.g., P‐selectin), promoting leukocyte–platelet aggregate formation [127, 128]. Treatment with evolocumab, a PCSK9 inhibitor, attenuated these prothrombotic effects by 60–70%, normalizing platelet reactivity. CD36 knockout models demonstrated that PCSK9's thrombogenic actions are CD36‐dependent, mediated through activation of the Src kinase/MAPK signaling axis (ERK5, JNK) and downstream pathways, such as ROS production, p38MAPK/cytosolic phospholipase A2 activation and cyclooxygenase‐1 (COX‐1)‐dependent thromboxane A2 (TXA2) synthesis. Notably, aspirin administration completely abolished PCSK9‐enhanced platelet activation and thrombosis in vivo, suggesting synergistic therapeutic potential when combining PCSK9‐i with antiplatelet agents.
PCSK9 deficiency in mice confers strong protection against thrombosis, underscoring its critical role in platelet‐driven thrombotic processes and supporting therapeutic strategies targeting the thrombotic microenvironment. Aspirin completely reverses PCSK9‐enhanced thrombosis, confirming the central involvement of the COX‐1/TXA2 pathway and highlighting the value of combination therapies. Unlike conventional antiplatelet agents (e.g., aspirin, clopidogrel), which increase bleeding risk, PCSK9 inhibition may allow more precise targeting of pathological thrombosis without compromising hemostasis. Future work should explore synergies between PCSK9‐i and emerging antithrombotic agents (e.g., hirudin derivatives or natural anticoagulant peptides) to enhance efficacy while limiting bleeding risks. Further investigation is also needed into the interplay between PCSK9, CD36, and oxidative stress pathways to uncover upstream regulatory mechanisms and identify biomarkers for patient stratification.
Immunomodulatory Effects
4.4
In vitro studies using macrophage and endothelial cell models demonstrate that PCSK9 in VSMCs is regulated through the TLR4–SAPK/JNK signaling pathway, a critical mediator of inflammatory and metabolic processes. VSMC‐derived PCSK9 reduces monocyte LDLR expression and impairs LDL‐C/LDLR‐mediated CCR2 expression on monocytes, a mechanism essential for monocyte recruitment and atherogenesis [47, 129]. This inhibition also elevates MHC class I molecule expression on tumor cells within the tumor microenvironment, facilitating CD8+ T cell recognition and tumor cell elimination [130, 131, 132].
In a pristane‐induced systemic lupus erythematosus (SLE) mouse model, pharmacological inhibition of PCSK9 significantly attenuated systemic inflammation, as evidenced by a 40–50% reduction in proinflammatory cytokines (IL‐17, TNF‐α) and increase in anti‐inflammatory TGF‐β levels (p < 0.01). Clinical validation in SLE patients revealed a strong positive correlation between circulating PCSK9 levels and CRP (p < 0.001), further implicating PCSK9 in systemic inflammatory cascades. These findings position PCSK9 as a novel therapeutic target for autoimmune disorders, with inhibition offering dual benefits—ameliorating both metabolic dysregulation and inflammation‐driven tissue damage [45].
PCSK9 exerts immunomodulatory effects beyond lipid metabolism by engaging innate and adaptive immune pathways. It activates the TLR4–SAPK/JNK signaling axis, which is frequently associated with chronic inflammation and autoimmune conditions. Moreover, PCSK9 downregulates monocyte CCR2, potentially attenuating leukocyte migration to inflamed tissues—a mechanism aligned with studies where CCR2 blockade ameliorates inflammation in atherosclerotic and autoimmune models.
Notably, PCSK9 upregulates MHC‐I expression in tumor cells, enhancing antigen presentation and supporting synergistic potential with immunotherapies in oncology. Its correlation with CRP in SLE underscores its relevance as an immune‐metabolic biomarker and therapeutic target.
Critical unresolved questions involve tissue‐specific mechanisms of PCSK9–immune interactions and its impact on regulatory T cells and tolerogenic dendritic cells. Future investigations should evaluate PCSK9‐i in IL‐17‐ or TNF‐α‐driven autoimmune pathologies, such as rheumatoid arthritis and psoriasis. Integrated multiomics approaches may elucidate novel lipid–immune crosstalk, advancing precision immunotherapies targeting PCSK9.
Clinical Translation and Trial Evidence
5
PCSK9‐i are now established as a cornerstone of intensive lipid‐lowering therapy in ASCVD. They consistently reduce LDL‐C levels by 59–77% and lower ischemic stroke risk by 21–27%, while demonstrating a favorable safety profile across diverse populations, including those with chronic kidney disease (CKD), diabetes, and East Asian ancestry (Table 2) [133, 134, 135, 136, 137]. Beyond these foundational benefits, emerging evidence supports their role in plaque regression and acute‐phase management, further expanding their clinical utility.
Efficacy in Lipid‐Lowering and Cardiovascular Outcomes (MACE)
5.1
Synergistic LDL‐C Suppression and Stroke Risk Mitigation
5.1.1
PCSK9‐i demonstrate robust efficacy in reducing LDL‐C levels and mitigating MACEs, particularly ischemic stroke. The FOURIER trial (evolocumab), a landmark study involving 27, 564 patients with established ASCVD, achieved a 59% reduction in LDL‐C and a 21% lower risk of ischemic stroke (HR 0.79, 95% CI 0.66–0.95), with sustained benefits persisting through extended follow‐up periods [49, 138]. This aligns with findings from the ODYSSEY Outcomes trial (alirocumab), which enrolled 18, 924 patients post‐ASC. Alirocumab reduced stroke incidence by 27% (HR 0.73, 95% CI 0.57–0.93), while exhibiting no increased risk of hemorrhagic stroke (HR 0.83, 95% CI 0.42–1.65). Notably, this cerebroprotective effect remained consistent across high‐risk subgroups, including patients with baseline LDL‐C ≥ 100 mg/dL or prior cerebrovascular events, over a median follow‐up of 2.8 years [134, 139].
The mechanistic basis for stroke protection may extend beyond LDL‐C reduction alone. PCSK9‐i modulate inflammation pathways (e.g., reduced IL‐6 and hsCRP levels) and stabilize atherosclerotic plaques, thereby attenuating thromboembolic events originating from carotid or intracranial arteries. This dual action—lipid‐lowering and plaque stabilization—positions PCSK9‐i as uniquely effective in cerebrovascular risk mitigation.
Long‐term efficacy in LDL‐C control was further substantiated by the ODYSSEY LONG TERM trial. Adjunctive alirocumab therapy added to maximally tolerated statins achieved sustained LDL‐C reductions of 62% versus placebo (p < 0.001) over 78 weeks [140]. Subsequent posthoc analyses revealed a 15% relative risk reduction in MACE (composite of CV death, MI, or stroke), underscoring the correlation between profound LDL‐C lowering and cardiovascular risk attenuation.
In patients with refractory hypercholesterolemia, the ODYSSEY COMBO I/II trials demonstrated that alirocumab added to high‐intensity statin plus ezetimibe therapy achieved incremental LDL‐C reductions of 45–50% from baseline. This highlights its role in addressing unmet needs in lipid management, particularly where conventional therapies fail to achieve guideline‐directed LDL‐C targets [141].
Exploratory Insights Into Pleiotropic Effects in Humans
5.2
Determinants of Response: From Genetic Dyslipidemias to Metabolic Comorbidities
5.2.1
Homozygous FH (HoFH), a genetic disorder characterized by markedly elevated LDL‐C levels from birth, predisposes individuals to early‐onset and progressive ASCVD. Early therapeutic intervention is crucial for mitigating cardiovascular risk; however, many patients fail to achieve guideline‐recommended LDL‐C targets.
In the TAUSSIG trial (NCT01624142) evaluating evolocumab, a 23% reduction in LDL‐C was observed among HoFH patients, with a favorable safety profile [142, 143, 144]. By contrast, the ORION‐5 randomized clinical trial (NCT03851705) assessing inclisiran in HoFH demonstrated that despite significant reductions in PCSK9 levels, inclisiran therapy did not significantly lower LDL‐C concentrations in the adult HoFH cohort [145]. Nonetheless, inclisiran exhibited good tolerability, with safety findings consistent with prior studies. Conversely, Part 1 of the ORION‐13 study indicated that over 1 year, inclisiran effectively reduced LDL‐C in adolescent HoFH patients and was well tolerated [146]. These results suggest inclisiran may serve as a potential adjunctive therapy for adolescents with HoFH and minimal residual LDLR activity.
Beyond LDL‐C reduction, PCSK9 inhibition therapy may favorably influence cholesterol clearance kinetics in heterozygous FH (HeFH). In CKD populations, subgroup analyses from the FOURIER‐CKD study revealed preserved cardiovascular risk reduction benefit and acceptable safety with evolocumab in patients with estimated glomerular filtration rate (eGFR) <60 mL/min/1.73 m^2^. Findings from the ODYSSEY DM‐INSULIN and DM‐DYSLIPIDEMIA trials demonstrated significant LDL‐C lowering with alirocumab in diabetic patients, without significant alteration in glycemic control [147, 148].
Time‐Sensitive Intervention: From Plaque Morphology to Guideline‐Directed Therapy
5.2.2
Acute‐phase intervention studies, such as the EVOPACS trial, validated the safety and feasibility of rapid lipid‐lowering postevent, supporting early intensive therapy. Imaging studies like Huygens and PACMAN‐AMI further demonstrated that acute‐phase PCSK9 inhibitor treatment stabilizes atherosclerotic plaques, reducing lipid‐rich necrotic core volume and enhancing fibrous cap thickness [149].
The 2022 American Heart Association (AHA) Scientific Statement advocates for early intensive lipid‐lowering therapy incorporating PCSK9‐i in extremely high‐risk ASCVD populations. Complementarily, the 2023 ESC/EAS Guidelines assign Class I recommendations for PCSK9 inhibitor initiation in high‐risk ASCVD patients failing to achieve LDL‐C targets despite maximally tolerated statin plus ezetimibe therapy [150].
The FITTER trial—a multicenter, randomized, double‐blind, placebo‐controlled study—assessed PCSK9 inhibitor efficacy in statin‐treated ACS patients with multivessel disease. This investigation demonstrated substantial reductions in LDL‐C concentrations alongside significant improvements in coronary microvascular function within this high‐risk cohort [151].
Exploratory and Analytical Research Breakthroughs
5.2.3
OSLER‐1/2 (evolocumab) showed that no new safety signals were identified during more than 7 years of follow‐up. The FOURIER‐OLE results showed that after 5.5 years of continuous treatment, LDL‐C remained below 30 mg/dL, and cardiovascular benefits increased with prolonged treatment duration [64]. Additionally, the HEYMANS study (a prospective registry enrolling adults from 12 European countries who initiated evolocumab treatment in routine clinical practice) demonstrated that evolocumab treatment was associated with sustained reductions in LDL‐C levels for up to 30 months, and that the persistence of evolocumab was high at both 12 and 30 months. Expanding the use of monoclonal antibodies such as evolocumab can improve LDL‐C control at the population level and confirm safety outcomes consistent with randomized controlled trials (RCTs) [152, 153, 154].
Lipid reduction and fibrous cap thickening directly mitigate plaque rupture risk, constituting the pathophysiological mechanism underlying reduced cardiovascular events. Regarding cerebrovascular benefits, the PACMAN‐AMI trial (NCT03067844) utilizing evolocumab demonstrated significant coronary plaque regression via serial intravascular ultrasound (IVUS) assessment after 52 weeks. This landmark study constitutes the first multimodal imaging verification that PCSK9 inhibitor–statin combination therapy induces substantial plaque regression in nonculprit coronary arteries [97, 155, 156, 157, 158]. At the lesion‐specific level, intensive lipid‐lowering yielded >twofold greater regression in high‐risk vulnerable plaques compared with whole‐vessel analyses, providing compelling evidence for targeted therapy in high‐risk cohorts [159]. For secondary stroke prevention, posthoc analyses of the discontinued SPIRE program (investigating bococizumab, which exhibited neutralizing antibody‐mediated immunogenicity) suggested potential reduction in recurrent stroke risk [160]. Concurrently, the ongoing ORION‐4 trial (NCT03705234) is evaluating inclisiran's siRNA‐mediated PCSK9 suppression to determine its impact on recurrent cerebrovascular events‐findings that may support biannual administration of this RNA therapeutic [161].
Neuroprotective Disconnect across Species: BBB Impermeability and Molecular Size Constraints
5.2.4
Despite demonstrating pronounced neuroprotection in preclinical models—including significant reductions in cerebral infarct volume and improved neurological deficit scores—these findings exhibit limited translatability to human stroke populations [162, 163]. Most experimental paradigms utilize young, healthy transgenic rodents subjected to induced cerebral ischemia, failing to recapitulate clinical comorbidities (e.g., diabetes mellitus, chronic hypertension) or heterogeneous stroke etiologies (e.g., cardioembolic, lacunar) prevalent in human cohorts [164, 165, 166]. Additionally, therapeutic intervention windows in animal studies (typically pre‐ or peri‐ischemic administration) diverge substantially from clinical realities of delayed treatment initiation [167].
BBB impermeability constitutes a fundamental translational barrier, potentially restricting central nervous system bioavailability. The substantial molecular mass (∼150 kDa) of monoclonal antibody‐based PCSK9‐i (e.g., evolocumab) precludes passive diffusion across the BBB, likely limiting direct neuroprotective effects [168, 169]. While novel RNA interference therapeutics (e.g., inclisiran) offer extended pharmacodynamic profiles, their anionic macromolecular structure similarly impedes BBB transit [170, 171].
Bridging this translational gap necessitates: integrated analyses of postmortem human neurovascular tissue; dedicated clinical trials in cerebrovascular disease populations; cross‐species comparative pharmacodynamic studies. These approaches will elucidate the therapeutic relevance of PCSK9 inhibition in stroke pathophysiology.
Clinical Applications and Protocol Development
6
Studies demonstrate that in‐hospital initiation of PCSK9‐i within 72 h of ACS rapidly reduces LDL‐C by 50–60%, improves lipid target attainment, and lowers 30‐day MACE risk by 28%, with no significant safety concerns [172]. The 2023 AHA/ASA Guidelines formalize this approach, recommending PCSK9‐i combined with maximally tolerated statins for very high‐risk ischemic stroke patients with LDL‐C ≥70 mg/dL. This recommendation synthesizes evidence from pivotal trials, including: FOURIER trial: 59% LDL‐C reduction and 21% ischemic stroke risk reduction with evolocumab, SPARCL study: validation of intensive lipid‐lowering for secondary stroke prevention, 2021 AHA/ASA update: emphasis on LDL‐C thresholds for advanced therapies.
Challenges, Unmet Needs, and Future Perspectives
7
Translational Gaps and Limitations in Current Models
7.1
Differential Anti‐Inflammatory Effects at Systemic versus Tissue Levels in Humans
7.1.1
Multiple clinical trials demonstrate that PCSK9‐i exert minimal or negligible effects on reducing circulating inflammatory markers, such as hs‐CRP [138, 173, 174, 175]. Despite achieving substantial reductions in LDL‐C of 55–60%, these agents were associated with only a modest, nonsignificant decrease in hs‐CRP of 3.2% (95% CI: −8.1 to +1.7%; p = 0.21) [176, 177, 178, 179, 180, 181]. This dissociation between LDL‐C lowering and anti‐inflammatory effects was further investigated in the large‐scale SPIRE trial. Utilizing proteomic analysis, a dedicated sub‐study comprehensively assessed 92 plasma inflammatory proteins, including key cytokines like IL‐6 and TNF‐α [36]. The findings revealed that these inflammatory markers failed to exhibit significant alterations following PCSK9 inhibitor treatment, despite pronounced LDL‐C reduction. These data collectively suggest that the primary mechanism underlying the antiatherosclerotic efficacy of PCSK9‐i is robust lipid lowering, rather than direct modulation of inflammatory pathways. This view is reinforced by results from the PROMISE study, which highlighted that patients at high inflammatory risk (defined by hs‐CRP ≥ 3 mg/L) experienced a significantly elevated risk of cardiovascular events, even when target LDL‐C levels were successfully achieved. Regarding the observed biological dissociation between hs‐CRP and LDL‐C, a mechanistic explanation proposes that hs‐CRP, an established inflammatory biomarker, predominantly reflects the activation status of the IL‐6 signaling pathway [182]. In contrast, LDL‐C reduction via PCSK9 inhibition is primarily mediated by the upregulation of LDLRs on hepatocytes. These represent distinct, nonoverlapping pathophysiological processes [183, 184].
In vivo analyses demonstrated significant localized anti‐inflammatory effects within atherosclerotic plaques. A prespecified PACMAN‐AMI trial sub‐study revealed that alirocumab, a PCSK9 inhibitor, elicited significant reductions in plaque macrophage infiltration and inflammatory cytokine expression (notably IL‐6 and TNF‐α). Concurrently, it enhanced plaque stability through attenuated lipid core volume and increased fibrous cap thickness [61, 97, 185, 186].Comparative histopathological evaluation of carotid endarterectomy specimens further established that PCSK9 inhibitor‐treated patients exhibited significantly lower intraplaque inflammatory mediator levels versus statin‐treated counterparts, despite comparable achievement of guideline‐recommended LDL‐C targets (<70 mg/dL) [187, 188, 189]. Carotid MRI vasculature imaging corroborated these findings, demonstrating PCSK9 inhibitor therapy significantly suppressed plaque neovascularization and reduced concentrations of instability‐associated biomarkers, including osteopontin and MMPs [190, 191, 192, 193, 194].
Analysis revealed a significant localized anti‐inflammatory effect in the resolution of myocardial inflammation. The EVACS I/II trial demonstrated that early administration of the PCSK9 inhibitor evolocumab accelerates myocardial inflammatory resolution following ACS and may attenuate adverse cardiac remodeling, despite unaltered systemic inflammatory biomarkers [195, 196, 197]. Serum PCSK9 concentrations exhibit a positive correlation with myocardial inflammation severity. Mechanistically, PCSK9‐i suppress local NF‐κB activation through blockade of the CAP1–PKCδ signaling axis, thereby mitigating myocardial ischemia–reperfusion injury [198].
PCSK9‐i elicit significant local anti‐inflammatory effects on both vascular endothelial cells and smooth smooth muscle cells. Specifically, they effectively downregulate LOX‐1 receptor expression, attenuate oxLDL‐induced endothelial cell apoptosis and monocyte adhesion, thereby ameliorating vascular endothelial function [63]. Furthermore, studies employing animal models have demonstrated that PCSK9‐knockout or antibody‐mediated inhibition reduces macrophage infiltration and MMP‐9 expression within AAAs, inhibiting the degradation of elastic fibers in the vascular wall [36]. These findings indicate potent localized protective actions of PCSK9 inhibition on vascular biology.
The tissue‐restricted effects may originate from several distinct molecular mechanisms. First, PCSK9‐i significantly upregulate LDLR expression on plaque‐resident macrophages, thereby facilitating oxLDL efflux and clearance. This process attenuates NLRP3 inflammasome priming—an effect contingent upon achieving supraphysiologic local drug concentrations within atherosclerotic lesions, a pharmacokinetic profile not attainable during systemic circulation [41]. Second, these inhibitors potentially mitigate mitochondrial oxidative stress in plaque endothelial cells via SIRT3 upregulation, subsequently suppressing downstream NF‐κB signal transduction [42, 67]. This differential effect may account for the superior reduction in stroke risk observed with PCSK9 inhibition in the FOURIER trial, despite achieving LDL‐C levels comparable to those attained with other LDL‐lowering therapies. Future studies should use advanced imaging techniques to link PCSK9 inhibitor therapy with inflammatory changes in atherosclerotic plaques [199]. Additionally, investigating the durability of these tissue‐specific effects within high‐risk subgroups (e.g., individuals with diabetes mellitus) is warranted. The observed dissociation between systemic and local anti‐inflammatory responses underscores the imperative to develop atherosclerosis‐specific biomarkers that extend beyond conventional blood‐based assays.
Unmet Need in Stroke Prevention: Subtype‐Specific Efficacy Challenges
7.1.2
Although PCSK9‐i have demonstrated definitive efficacy in reducing MACEs, their impact on stroke‐related outcomes remains incompletely elucidated. Large‐scale RCTs, namely FOURIER (evolocumab) and ODYSSEY OUTCOMES (alirocumab), were primarily powered to assess composite cardiovascular endpoints, incorporating stroke as a secondary or exploratory component [25, 27, 86]. Subsequent subgroup analyses within these trials have suggested a trend toward stroke risk reduction; however, these findings frequently lack statistical significance or have demonstrated marginal clinical impact relative to the primary outcome. This attenuation of effect on stroke prevention may be attributable, in part, to specific design limitations inherent in the existing trials.
Current clinical evidence on PCSK9‐i predominantly focuses on coronary artery disease populations, with limited representation of high‐risk stroke cohorts. Notably, only 15–20% of participants in major trials had a prior history of cerebrovascular events, while critical subgroups such as intracranial atherosclerotic stenosis (ICAS) patients—a population with inherently elevated stroke vulnerability—remain markedly underrepresented. This gap underscores a significant limitation in extrapolating cardiovascular outcomes to neurovascular prevention paradigms. The landmark 2025 ICAS prospective cohort study addressed this research void, demonstrating that adjunctive PCSK9 inhibitor therapy (combined with statins) achieved a 66.5% reduction in early recurrent stroke risk. Critically, the LDL‐C reduction was significantly more pronounced in the combination group, establishing a robust dose–response relationship between lipid‐lowering intensity and secondary stroke prevention efficacy in ICAS [200]. Second, the follow‐up durations in these trials, typically 2–3 years, may be insufficient to reliably detect a significant preventive effect on ischemic stroke. This timeframe could potentially be inadequate for manifesting the full impact of LDL‐C lowering via PCSK9‐i on atherosclerotic plaque stabilization and regression within the cerebrovascular circulation, processes hypothesized to evolve over a more protracted timeline than the stabilization observed in ACSs [201].
To date, no large‐scale RCTs have performed stratified outcome analyses based on stroke etiologic subtypes (TOAST classification) [173, 202]. Existing studies have failed to demonstrate a significant preventive efficacy of PCSK9 inhibitors for either cardioembolic or small‐vessel occlusion (lacunar) stroke. Pooled analyses indicate that PCSK9 inhibition may lower the risk of large‐artery atherosclerotic stroke—an effect attributable to its LDL‐C‐lowering mechanism—but exhibits no discernible effect on cardioembolic or small‐vessel strokes. Furthermore, persistent concerns remain that achieving very low LDL‐C concentrations (<25 mg/dL [<0.65 mmol/L]) may elevate hemorrhagic stroke risk, a signal identified in the FOURIER trial [114]. However, the limited number of events precludes definitive conclusions regarding this potential association.
In conclusion, while there is biological plausibility supporting a role for PCSK9‐i in stroke prevention, definitive evidence of clinically significant benefit requires dedicated outcome trials with prolonged follow‐up enrolling high‐risk cohorts. Presently, the primary indication for these agents remains cardiovascular risk reduction, with insufficient evidence to support a stroke‐specific protective effect.
The role of PCSK9‐i in stroke prevention necessitates a paradigm shift from a “lipid‐lowering‐centric” approach to a “multimodal synergistic” strategy. Current evidence supports significant benefits in specific high‐risk populations (e.g., symptomatic ICAS or elevated lipoprotein(a) [Lp(a)] levels), yet their ineffectiveness in nonatherosclerotic strokes (such as cardioembolic events) underscores the need for individualized therapy. Future research should focus on validating long‐term safety and clinical translational value of their pleiotropic effects through dedicated RCTs, while concurrently developing predictive models to optimize treatment cost effectiveness.
Limitations of PCSK9‐i in Translating to Human Stroke Pathophysiology
7.1.3
Current evidence does not establish a cerebrovascular‐specific mechanism for PCSK9‐i in human stroke. Research on PCSK9 in atherosclerosis primarily stems from coronary or peripheral vascular studies. However, the cerebrovascular microenvironment—including the BBB and local inflammation—may differ significantly [203]. Regarding carotid plaque mechanisms, studies ex vivo using human carotid plaques demonstrate that PCSK9 induces mitochondrial fission via the p38–DRP1 pathway, promoting VSMC apoptosis and plaque vulnerability. This finding lacks direct validation in cerebral arteries [55, 204]. PCSK9 also exacerbates cerebrovascular endothelial inflammation and oxidative stress via the LOX‐1 receptor, but whether this regulatory mechanism aligns with coronary arteries remains unclear [205]. Future research should address these gaps through cross‐organ cellular studies, molecular analyses of intracranial plaques, and targeted animal models to determine the pathway's universality and therapeutic potential in cerebral arterial disease.
Regarding translational gaps in neuroprotection, animal studies suggest PCSK9‐i may attenuate postischemic neuronal apoptosis by downregulating ApoER2. However, human stroke involves more complex neuroinflammation and glial responses, while existing studies have not distinguished between stroke subtypes (e.g., embolic versus thrombotic) [124, 206, 207]. Future research requires molecular subtyping studies across stroke subtypes, integrated with multiomics approaches (e.g., single‐cell sequencing) to elucidate the regulatory networks of neuroinflammation and glial responses, thereby advancing precision neuroprotective strategies.
A key limitation is the lack of robust human data demonstrating a direct effect of PCSK9‐i on stroke‐specific outcomes. Although large cardiovascular outcome trials (e.g., FOURIER, ODYSSEY) reported reductions in composite endpoints including stroke, they were not designed to isolate stroke mechanisms or assess neuroprotective effects. Furthermore, no definitive evidence currently demonstrates PCSK9 inhibitor efficacy in hemorrhagic stroke, and theoretical concerns persist that very low LDL‐C levels (<30 mg/dL) may increase intracranial hemorrhage risk—a signal noted in some trials requiring further investigation. Future research necessitates randomized trials with neuroprotection as a primary endpoint, integrated with multiomics approaches for mechanistic insights.
The mechanisms by which PCSK9‐i affect stroke pathophysiology require further clinical validation. While the multiple effects of PCSK9‐i (e.g., anti‐inflammatory and antioxidant properties) show promise in vitro and in animal models, conclusive human evidence is still lacking. Some patients exhibit reduced plaque lipid core post‐PCSK9 inhibitor administration without improved inflammatory markers, suggesting insignificant anti‐inflammatory effects [138, 208]. LOF mutations (e.g., R46L) lower LDL‐C and ischemic stroke risk, but distinguishing direct cerebrovascular protection from systemic lipid‐lowering effects remains challenging [209, 210, 211].
Research reliance on surrogate endpoints (e.g., cIMT, cfPWV) rather than direct stroke outcomes highlights biomarker limitations [212, 213]. The lack of dedicated stroke trials evaluating imaging biomarkers (e.g., carotid MRI‐assessed plaque stability) or mechanistic endpoints (e.g., microglial activation) impedes clinical translation [214, 215, 216]. Bridging this gap requires multiomics data integration (e.g., metabolomics + proteomics + imaging) via machine learning and adaptive trial designs (e.g., umbrella trials) to validate composite endpoints.
Novel Therapeutic Technologies and Drug Development
7.2
Small‐Molecule PCSK9 Antagonism: Clinical‐Stage Oral Delivery Advancement
7.2.1
AZD0780 (AstraZeneca), a small molecule targeting PCSK9–LDLR interactions, demonstrated dose‐dependent efficacy in the phase II PURSUIT trial. At the 30 mg dose combined with statins, it achieved a 50.7% reduction in LDL‐C over 12 weeks, highlighting its potential for integration into conventional lipid‐lowering regimens [217]. MK‐0616, another oral small molecule inhibitor, has shown promising results across early trials: phase I data confirmed its safety and tolerability, while phase IIb trials revealed a 60.9% LDL‐C reduction at the 30 mg dose. This robust efficacy has propelled MK‐0616 into global phase III clinical testing, positioning it as a frontrunner in oral PCSK9 inhibition [218, 219].
Simultaneously, CVI‐LM001, the first‐in‐class small molecule inhibitor targeting hepatocyte nuclear factor‐1α to indirectly modulate PCSK9 expression, has entered phase II trials. Its unique mechanism bypasses direct PCSK9 binding, potentially reducing off‐target effects while maintaining significant LDL‐lowering activity [218].
Epigenome Editing for Sustained Silencing: Lipid Nanoparticle‐Delivered Methylation Therapy
7.2.2
PCSK9‐EE (developed by Chroma Medicine et al.) utilizes lipid nanoparticles to deliver an epigenetic editor—a DNMT3A/dCas9 complex—that specifically targets the PCSK9 promoter region. This innovative approach induces DNA methylation at the promoter site, achieving durable gene silencing and sustained LDL‐C reduction. In preclinical studies, a refined candidate, PCSK9‐EE‐V2, demonstrated optimal activity and persistence in nonhuman primates, maintaining >70% PCSK9 suppression for over 12 months posttreatment. Further optimization of delivery specificity and safety profiles is ongoing to advance clinical translation [167].
Adherence‐Optimized Synergy: Infrequent Dosing With Complementary Mechanisms
7.2.3
The development of long‐acting siRNA therapies, such as inclisiran, has opened new avenues for synergistic lipid management (Figure 4). When combined with statins or oral PCSK9‐i, inclisiran demonstrates the potential to achieve LDL‐C reductions exceeding 80%, leveraging its biannual dosing regimen to sustain profound lipid‐lowering effects. For instance, the AZD0780 (oral PCSK9 inhibitor) and rosuvastatin combination, evaluated in a phase I trial, reduced LDL‐C by an additional 51% compared with statin monotherapy, highlighting the potent additive effects of dual‐mechanism approaches. Emerging data suggest such combinations not only enhance efficacy but may also improve medication adherence by reducing dosing frequency. However, long‐term safety and cost effectiveness of these regimens require further validation in larger, diverse patient populations [217].
Mechanism of action of inclisiran. Inclisiran is a first‐in‐class synthetic small‐interfering RNA (siRNA) therapeutic designed to specifically target and silence the PCSK9 gene in hepatocytes. Its mechanism involves a sophisticated RNA interference (RNAi) pathway: after subcutaneous administration, inclisiran is transported to the liver where it binds to the RNA‐induced silencing complex (RISC). The activated RISC then selectively cleaves PCSK9 messenger RNA (mRNA), preventing its translation into functional PCSK9 protein. By inhibiting PCSK9 synthesis at the genetic level, inclisiran upregulates hepatic LDL receptors (LDLR) that would otherwise be degraded by PCSK9. This leads to enhanced LDLR recycling on hepatocyte surfaces, significantly improving LDL‐cholesterol (LDL‐C) clearance from the bloodstream. The RNAi‐mediated effect provides prolonged pharmacological activity, with a single dose achieving >50% reduction in circulating PCSK9 and LDL‐C levels for 3–6 months.
Disruptive Therapeutic Platforms: Base Editors and Antisense Oligonucleotides
7.2.4
VERVE‐101, a pioneering base‐editing therapy, achieves permanent PCSK9 gene knockout in hepatocytes, demonstrating robust LDL‐C reduction (60%) in early animal trials (Figure 5). While promising, challenges persist in optimizing liver‐specific delivery and ensuring long‐term safety, particularly regarding off‐target genomic effects and immune responses to editing components [167]. Concurrently, pelacarsen, an antisense oligonucleotide targeting Lp(a), has shown remarkable efficacy in Phase II trials, reducing Lp(a) levels by 72–80% through selective degradation of apolipoprotein(a) [apo(a)] mRNA. This reduction correlates with attenuated inflammation and thrombotic risk, positioning pelacarsen as a potential game‐changer for patients with elevated Lp(a)‐driven cardiovascular risk [220].
Mechanism of action of VERVE‐101. VERVE‐101 is an innovative base‐editing therapy designed to permanently inactivate the PCSK9 gene in hepatocytes, offering a potential one‐time treatment for hypercholesterolemia. This CRISPR‐based therapy utilizes an adenine base editor (ABE) delivered via lipid nanoparticles (LNPs) to precisely convert a single adenine (A) to guanine (G) in the PCSK9 gene, creating a premature stop codon that halts PCSK9 protein production. By permanently silencing PCSK9 at the DNA level, VERVE‐101 upregulates hepatic LDL receptors (LDLR), mimicking natural loss‐of‐function mutations associated with lifelong low LDL‐C. Unlike RNAi therapies (e.g., inclisiran) requiring redosing, VERVE‐101's single‐administration approach enables durable PCSK9 knockout, potentially providing lifelong LDL‐C reduction (>50%) and cardiovascular protection. The LNP delivery system ensures hepatocyte‐specific targeting, minimizing off‐target effects. Preclinical data show efficient PCSK9 editing (>80%) with sustained LDL‐C lowering.
Defining Future Research Agendas
7.3
Multidimensional Knowledge Gaps: Therapeutic Innovation Barriers
7.3.1
Key knowledge gaps include defining the optimal dosing windows and dose optimization during acute‐phase treatment, as well as clarifying the long‐term safety profile, particularly regarding cognitive function impacts with sustained ultra‐low LDL‐C levels (<20 mg/dL). Advancing novel formulations—such as oral PCSK9‐i or ultra‐long‐acting injectables—remains critical to improve adherence, while emerging approaches like PCSK9 vaccines and gene‐editing therapies (e.g., VERVE‐101) aim to achieve durable benefits through single‐course interventions.
Precision medicine strategies are being developed to identify high‐risk patients through predictive models incorporating ALT levels, baseline LDL‐C, and statin responsiveness. Concurrently, combination therapies targeting residual risks—such as pairing PCSK9‐i with Lp(a)‐lowering RNA therapies (e.g., pelacarsen)—are under investigation to address multifaceted lipid and inflammatory pathways.
Basic research priorities include elucidating the role of PCSK9 in acute ischemic stroke and intracranial artery stenosis, as well as exploring synergistic regimens combining PCSK9‐i with statins and antiplatelet agents (e.g., aspirin) to achieve multiple effects through lipid‐lowering, anti‐inflammatory, and antithrombotic effects. Indication expansion efforts focus on clarifying PCSK9's platelet CD36 receptor interactions and nonlipid pathways to develop next‐generation therapies targeting novel mechanisms (e.g., CAP1 inhibition). Large‐scale RCTs are urgently needed to validate PCSK9‐i’ efficacy in primary prevention of VTE and other noncardiovascular indications. Addressing these challenges will require interdisciplinary collaboration to translate mechanistic insights into transformative clinical applications.
Resource Allocation Efficiency: Cost‐Accessibility Disparities in Stroke Prophylaxis
7.3.2
Despite the demonstrated efficacy of PCSK9‐i in reducing LDL‐C and cardiovascular events, their widespread adoption for stroke prevention faces significant financial and operational barriers. High treatment cost remains the primary obstacle, with annual per‐patient expenses substantially exceeding those of generic statins [221, 222, 223]. Cost‐effectiveness debates persist, and the high cost drives stringent prior authorization requirements (US rejection rates: 40–60%), increasing administrative burdens [224, 225].
Accessibility challenges further impede implementation. Utilization is highly inequitable, with over 80% concentrated in high‐income countries. Low‐ and middle‐income countries face prohibitive costs and lack infrastructure for monoclonal antibody injection, while remote areas encounter additional barriers [226, 227, 228]. Patient adherence is suboptimal due to the requirement for biweekly subcutaneous injections compared with oral therapies [229].
Innovative solutions are being explored. Stroke guidelines recommend PCSK9‐i use concomitant with healthcare payment reforms to ensure equitable access. Current economic thresholds restrict PCSK9‐i use primarily to high‐risk patients within well‐resourced systems, despite their therapeutic potential. Future research should focus on: real‐world data‐based pharmacoeconomic evaluations in stroke‐specific populations and developing risk‐stratification tools to optimize targeted therapy.
Conclusion
8
PCSK9‐i have demonstrated significant therapeutic potential across the spectrum of ischemic stroke management, encompassing primary prevention, secondary prevention, acute‐phase treatment, and long‐term prognosis improvement. Their multifaceted mechanisms of action—including LDL‐C reduction, stabilization of atherosclerotic plaques via anti‐inflammatory effects, attenuation of thrombotic risk, and endothelial function restoration—position these agents as versatile tools for addressing both the metabolic and inflammatory drivers of cerebrovascular disease. Clinical trials such as FOURIER and ODYSSEY Outcomes have validated their capacity to reduce ischemic stroke risk by 21–27% while maintaining a favorable safety profile, even in high‐risk populations with comorbid conditions.
Despite these advances, limitations persist in current research and clinical application. The long‐term cognitive effects of sustained LDL‐C lowering to very low levels (<20 mg/dL) remain under investigation, and optimal timing for acute‐phase intervention requires further refinement. Additionally, cost effectiveness and accessibility challenges hinder widespread adoption in resource‐limited settings. Future high‐quality studies are needed to elucidate the full scope of PCSK9‐i’ multiple benefits, establish standardized treatment protocols for vulnerable plaque phenotypes, and explore synergistic effects with emerging therapies like antiplatelet agents or Lp(a)‐targeted drugs.
With ongoing research into novel formulations (e.g., oral PCSK9‐i, siRNA therapies) and biomarker‐guided approaches, PCSK9‐i are poised to become cornerstone therapies in ischemic stroke management. Their unique ability to concurrently target lipid metabolism, vascular inflammation, and thrombotic pathways offers unprecedented opportunities to improve clinical outcomes and quality of life for stroke survivors, ultimately reducing the global burden of cerebrovascular disease.
Simultaneously, the documented adverse effect profile of PCSK9‐i warrants consideration. While these agents lack association with statin‐class adverse effects such as hepatic dysfunction, new‐onset diabetes mellitus, rhabdomyolysis, or significant ophthalmic complications, they may elicit localized or systemic reactions. These include injection site discomfort (e.g., myalgia), cephalgia, and influenza‐like symptoms. Furthermore, although certain investigations suggest potential associations with increased hemorrhagic risk and altered neurocognitive function, the current evidence base remains insufficient. Definitive confirmation of these relationships necessitates further prospective, large‐scale studies.
As the therapeutic evidence evolves, PCSK9‐i demonstrate significant potential as an adjunctive or alternative therapeutic strategy in the secondary prevention of ischemic stroke. Their integration into management algorithms holds promise for substantially improving clinical outcomes in this patient population.
Author Contributions
Jia Kuang and Lei Hao participated in writing. Zhao Yang participated in reviewing and editing. All authors have read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Ethics Statement
No ethical approval was required for this study.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1D. Sims , “Ask the Consultant: Stroke, ” Bmj 389 (2025): r 436.40246315 10.1136/bmj.r 436 · doi ↗ · pubmed ↗
- 2V. L. Feigin , M. Brainin , B. Norrving , et al., “World stroke organization (WSO): Global stroke Fact Sheet 2022, ” International Journal of Stroke 17 (2022): 18–29.34986727 10.1177/17474930211065917 · doi ↗ · pubmed ↗
- 3F. Mach , C. Baigent , A. L. Catapano , et al., “2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk, ” European Heart Journal 41 (2020): 111–188.31504418 10.1093/eurheartj/ehz 455 · doi ↗ · pubmed ↗
- 4P. Gargiulo , C. Basile , A. Cesaro , et al., “Efficacy, Safety, Adherence and Persistence of PCSK 9 Inhibitors in Clinical Practice: A Single Country, Multicenter, Observational Study (AT‐TARGET‐IT), ” Atherosclerosis 366 (2023): 32–39.36696749 10.1016/j.atherosclerosis.2023.01.001 · doi ↗ · pubmed ↗
- 5V. L. Feigin , B. A. Stark , C. O. Johnson , et al., “Global, Regional, and National Burden of Stroke and Its Risk Factors, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019, ” Lancet Neurology 20 (2021): 795–820.34487721 10.1016/S 1474-4422(21)00252-0PMC 8443449 · doi ↗ · pubmed ↗
- 6J. Vicente‐Valor , X. García‐González , S. Ibáñez‐García , et al., “PCSK 9 inhibitors Revisited: Effectiveness and Safety of PCSK 9 inhibitors in a Real‐life spanish Cohort, ” Biomedicine and Pharmacotherapy 146 (2022): 112519.34968928 10.1016/j.biopha.2021.112519 · doi ↗ · pubmed ↗
- 7X. Bao , Y. Liang , H. Chang , et al., “Targeting Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK 9): From Bench to Bedside, ” Signal Transduction and Targeted Therapy 9 (2024): 13.38185721 10.1038/s 41392-023-01690-3PMC 10772138 · doi ↗ · pubmed ↗
- 8H. H. Hobbs , J. C. Cohen , and J. D. Horton , “PCSK 9: From Nature's Loss to Patient's Gain, ” Circulation 149 (2024): 171–173.38227713 10.1161/CIRCULATIONAHA.123.064498 PMC 10874118 · doi ↗ · pubmed ↗