Uncommon Origin, Common Presentation: A Rare Case of Mesenteric Carcinoid Tumor Mimicking Functional Gastrointestinal Disorders
Radhika Mathur, Rahul Borra, Salman Muddassir

TL;DR
A rare case of a mesenteric carcinoid tumor was mistaken for common GI issues, highlighting the need for early detection in atypical presentations.
Contribution
Presents a rare case of a mesenteric carcinoid tumor with atypical symptoms and emphasizes the importance of early recognition.
Findings
A 59-year-old woman presented with GI symptoms initially misdiagnosed but later found to have a mesenteric carcinoid tumor.
Elevated chromogranin A and biopsy confirmed a grade 1 neuroendocrine tumor with hepatic metastases.
The case underscores the challenge of diagnosing carcinoid tumors due to their nonspecific symptoms.
Abstract
Carcinoid tumors, a subtype of well-differentiated neuroendocrine neoplasms, are frequently misdiagnosed due to their indolent course and nonspecific gastrointestinal (GI) symptoms that mimic more common disorders. Here we report a rare case of a 59-year-old woman with hepatitis C and a history of intravenous drug use who presented with progressive abdominal pain, nausea, vomiting, diarrhea, and weight loss. Initial evaluations were inconclusive, but further imaging revealed a central mesenteric mass-an uncommon primary site-along with hepatic lesions, raising concern for metastatic disease. Elevated chromogranin A and biopsy confirmed a grade 1 neuroendocrine tumor. This case highlights the importance of considering carcinoid tumors in patients with persistent GI symptoms and emphasizes early recognition, particularly in atypical presentations, to guide timely and effective management.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
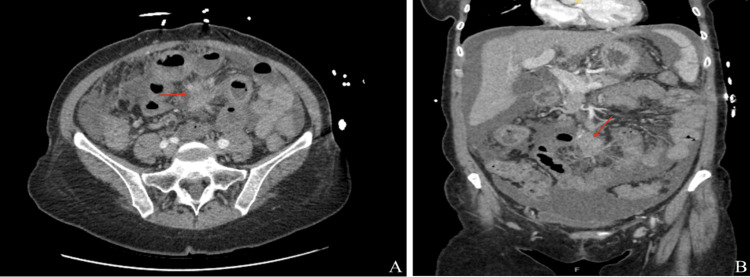 Figure 1
Figure 1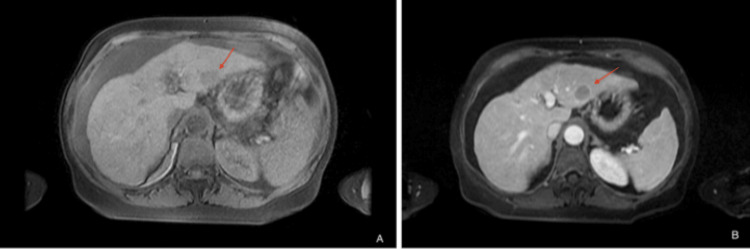 Figure 2
Figure 2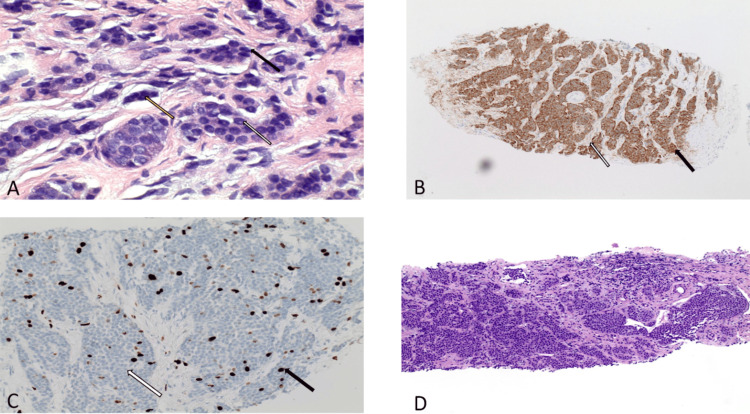 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeuroendocrine Tumor Research Advances · Neuroblastoma Research and Treatments · Pancreatic and Hepatic Oncology Research
Introduction
Carcinoid tumors are well-differentiated neuroendocrine neoplasms (NETs) that arise from enterochromaffin cells and most commonly originate in the gastrointestinal tract-particularly the ileum, appendix, rectum, and stomach. While the bronchopulmonary system is the second most frequent site for NETs, primary mesenteric tumors are exceptionally rare, reported only in isolated cases [1,2], with most mesenteric involvement representing secondary disease. Population registries do not list mesenteric primaries separately, underscoring their rarity [3,4].
Despite their typically indolent behavior, carcinoid tumors possess malignant potential and frequently secrete bioactive substances, causing nonspecific symptoms such as abdominal pain, diarrhea, flushing, and weight loss [5]. These manifestations often mimic more common functional or inflammatory gastrointestinal disorders, which can lead to misdiagnosis and delayed recognition [6]. The incidence of carcinoid tumors has risen significantly over recent decades due to heightened clinical awareness and advances in diagnostic modalities. Serum chromogranin A remains a useful biochemical marker, while CT, MRI, and somatostatin receptor PET/CT provide valuable information for localization and staging [7,8]. Histopathological evaluation remains essential, with tumor grade and Ki-67 index serving as critical prognostic indicators [9]. Although slow-growing, carcinoid tumors frequently metastasize to the liver, significantly impacting prognosis and management strategies. Treatment options include surgical resection when feasible, somatostatin analogs for tumor stabilization and symptom control, and systemic therapies in advanced or progressive disease [10,11].
Of note, tumors in unusual sites like the mesentery pose a diagnostic challenge, as extraluminal lesions often lack early obstructive symptoms and can mimic desmoid tumors, lymphoma, or metastases on imaging [12]. Furthermore, in patients with comorbidities such as treated hepatitis C, overlapping hepatic-related complaints-including abdominal discomfort, fatigue, or abnormal liver imaging-can obscure the clinical picture and delay recognition of a neuroendocrine tumor. This overlap highlights the need for a high index of suspicion and a systematic approach to distinguishing hepatic pathology related to chronic viral infection from metastatic disease arising from mesenteric NETs.
This case underscores the importance of considering carcinoid tumors in the differential diagnosis of persistent or unexplained gastrointestinal symptoms, particularly when extraluminal mesenteric lesions are identified. It also illustrates how concurrent conditions like hepatitis C may confound the diagnostic pathway [13], emphasizing the critical role of histopathology and comprehensive imaging in reaching a timely and accurate diagnosis.
Case presentation
A 59-year-old woman with a medical history significant for intravenous drug use and successfully treated hepatitis C presented to the emergency department with a two-month history of progressively worsening, crampy, generalized abdominal pain. Her symptoms were accompanied by persistent nausea, intermittent vomiting, watery diarrhea, early satiety, and a 20-pound unintentional weight loss. She denied hematochezia, melena, fevers, chills, or jaundice. Her physical examination was notable for diffuse abdominal tenderness without rebound or guarding, mild distention, and trace pedal edema. Vital signs were within normal limits.
Given the vague and nonspecific nature of her symptoms, initial clinical impressions included chronic liver disease, functional bowel disorder, or occult infection. She had undergone a colonoscopy nine years earlier that was unremarkable, and there was no family history of gastrointestinal malignancy. Basic laboratory evaluation was non-revealing, with normal white blood cell count, liver enzymes, and lipase. However, given the persistence and severity of her symptoms, particularly the significant weight loss, cross-sectional imaging was pursued.
Contrast-enhanced computed tomography (CT) of the abdomen and pelvis revealed an ill-defined central mesenteric soft tissue mass measuring 2.1 × 2.2 × 3.6 cm (Figures 1A, 1B), associated with radiating fibrotic strands and tethering of adjacent small bowel loops. The imaging appearance was suspicious for a desmoplastic reaction, raising concern for a carcinoid tumor. Other differential considerations included desmoid tumor, lymphoma, or metastatic disease. Additional findings included omental stranding, moderate ascites, cholelithiasis, common bile duct dilation up to 12 mm, and a nodular liver contour suggestive of underlying cirrhosis.
Axial (A) and sagittal (B) views of CT abdomen/pelvis demonstrating an ill-defined mesenteric soft tissue mass
At this point, the diagnosis remained uncertain. The central location of the mass in the mesentery, rather than the bowel wall, further complicated the identification of a primary tumor site, making this an unusual presentation. Gastroenterology was consulted and recommended further evaluation with tumor markers, including alpha-fetoprotein (AFP) and chromogranin A, as well as magnetic resonance imaging (MRI) of the abdomen and magnetic resonance cholangiopancreatography (MRCP). MRI/MRCP confirmed the presence of the liver metastasis (Figure 2A) and revealed multiple arterially enhancing sub-centimeter hepatic lesions, the largest measuring 4 mm (Figure 2B).
T1 fat saturated sequence showing ill-defined mass in the left hepatic lobe (A), followed by arterial enhancing mass in the left hepatic lobe measuring 4 mm without washout or capsule (B)
AFP was within normal range (3.6 ng/mL), while chromogranin A was markedly elevated at 114.9 ng/mL, heightening suspicion for a neuroendocrine tumor.
CT-guided biopsy of the mesenteric mass revealed cords and nests of small, uniform cells with increased nuclear-to-cytoplasmic ratios (Figure 3A) and characteristic “salt-and-pepper” chromatin. Immunohistochemistry demonstrated positivity for neuroendocrine markers CD56 and synaptophysin (Figure 3B), with a low Ki-67 proliferation index (Figure 3C) consistent with a grade 1 well-differentiated neuroendocrine tumor. CDX2 staining suggested a lower gastrointestinal origin. Biopsy of a hepatic lesion confirmed metastatic disease with identical histopathology (Figure 3D).
(A) Higher power demonstrating uniform cells with increased nuclear to cytoplasmic ratio (black arrow), smudged chromatin (yellow arrow) and inconspicuous nucleoli (white arrow); (B) synaptophysin immunohistochemical stain which is another marker of neuroendocrine differentiation (black arrow), fibrous septa (white arrow); (C) overview image of Ki- 67 staining where brown stain indicates actively dividing cells (black arrow), while blue staining (usually from hematoxylin) indicates cells that are not actively dividing (white arrow); (D) biopsy from the liver showing metastatic well-differentiated neuroendocrine tumor
The patient was diagnosed with a metastatic grade 1 well-differentiated neuroendocrine tumor (carcinoid tumor) of probable lower GI origin, presenting with a primary mesenteric mass, a rare and often under-recognized site. Given the low Ki-67 index, indicative of a slow-growing, low-grade tumor with limited expected benefit from systemic chemotherapy, she was initiated on subcutaneous octreotide (Sandostatin) 100 mcg twice daily for symptom control and tumor stabilization. Palliative care consultation and close outpatient surgery and oncology follow-up were also recommended to optimize symptom management and guide long-term monitoring of her indolent disease.
Discussion
Carcinoid tumors are slow-growing neuroendocrine neoplasms that frequently present with vague gastrointestinal complaints, often mimicking more common conditions and leading to diagnostic delays [3]. This case of a 59-year-old woman with a history of intravenous drug use, treated hepatitis C, and chronic, nonspecific gastrointestinal symptoms underscores the importance of maintaining a high index of suspicion for carcinoid tumors-particularly when imaging identifies a mesenteric mass with concurrent hepatic lesions. Extraluminal mesenteric tumors may not cause early obstructive symptoms, and their radiologic features can overlap with other pathologies such as desmoid tumors, lymphoma, or metastatic disease, complicating diagnosis [12]. In this context, underlying liver disease related to hepatitis C may obscure recognition of hepatic metastases, creating an additional diagnostic challenge. Nevertheless, liver metastases are common in carcinoid tumors and have a significant impact on both prognosis and management [8].
A review of the literature highlights both the rarity and complexity of this presentation. While carcinoid tumors most commonly arise in the small intestine, appendix, rectum, or stomach, primary mesenteric carcinoid tumors are exceptionally rare, reported only in isolated cases [1,2]. Chronic hepatitis C has been associated with gastric and small intestinal carcinoid tumors, suggesting that long-standing liver disease may indirectly influence tumor development or detection [13]. Although direct associations with intravenous drug use are limited, its link to immunocompromised states may increase susceptibility to malignancies, including NETs. Taken together, these factors underscore the diagnostic difficulty in patients with overlapping comorbidities and atypical tumor sites.
Diagnosis should follow a systematic, guideline-based approach. Biochemical evaluation, including serum chromogranin A and 24-hour urinary 5-hydroxyindoleacetic acid (5-HIAA), is recommended to support the suspicion of a neuroendocrine tumor and to monitor disease progression [4]. Cross-sectional imaging with contrast-enhanced CT or MRI of the abdomen and pelvis is essential for localizing the primary tumor and evaluating for mesenteric or hepatic involvement. Functional imaging, such as somatostatin receptor PET/CT, further refines staging and treatment planning. Histopathological confirmation through image-guided biopsy remains the gold standard, as demonstrated in this case by a CT-guided biopsy confirming a well-differentiated, grade 1 neuroendocrine tumor. The Ki-67 proliferative index provides key prognostic information and guides therapeutic decision-making; in this patient, a low Ki-67 index (<3%) reflected an indolent course with anticipated responsiveness to somatostatin analog therapy.
Therapeutically, somatostatin analogs such as octreotide remain the cornerstone of management for symptom control and disease stabilization in metastatic carcinoid tumors [9]. Current National Comprehensive Cancer Network (NCCN) and European Neuroendocrine Tumor Society (ENETS) guidelines recommend somatostatin analogs as first-line therapy for unresectable or metastatic small intestinal and mesenteric NETs [14,15]. In cases of disease progression, additional treatment modalities, including peptide receptor radionuclide therapy (PRRT), everolimus, or liver-directed interventions, should be considered.
This case highlights the critical need to consider carcinoid tumors in patients with persistent, unexplained gastrointestinal symptoms and suspicious imaging findings. Prognosis is strongly influenced by tumor grade, stage, and proliferative index. For mesenteric NETs specifically, survival outcomes are less favorable than for other gastrointestinal NETs: large series report five-year overall survival ranging from ~46-75% in patients with metastatic small intestinal NETs with mesenteric involvement, with significantly reduced survival when large mesenteric tumor deposits are present (65.5% vs. 92.6% without deposits), and a median survival of approximately 52 months [11].
Conclusions
This case underscores the diagnostic challenge posed by neuroendocrine tumors, especially when presenting with nonspecific gastrointestinal symptoms and the exceptionally rare primary mesenteric location. A history of hepatitis C and intravenous drug use could have further complicated disease progression and recognition, reflecting how comorbidities may obscure the clinical picture. Early diagnosis through guideline-directed biochemical, radiologic, and histopathologic evaluation is crucial, as hepatic metastases are common and carry significant prognostic weight. Prompt initiation of somatostatin analog therapy, particularly in low-grade tumors, can stabilize disease and improve quality of life. Clinicians should therefore maintain a high index of suspicion for neuroendocrine neoplasms in patients with persistent, unexplained GI complaints, even when alternative explanations seem likely.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Primary mesenteric carcinoid tumor J Korean Surg Soc Park IS Kye BH Kim HS Kim HJ Cho HM Yoo C Hwang SS 1141178420132339704410.4174/jkss.2013.84.2.114PMC 3566469 · doi ↗ · pubmed ↗
- 2Primary mesenteric neuroendocrine tumor: case report Int J Surg Case Rep González-Muñoz A Aguirre-Salamanca EJ Rivera-Rincón NA Rodríguez-Narvaez JG González-Sierra P Ramírez-Giraldo C 10851710920233750652910.1016/j.ijscr.2023.108517 PMC 10403702 · doi ↗ · pubmed ↗
- 3NANETS consensus guidelines for the diagnosis of neuroendocrine tumor Pancreas Vinik AI Woltering EA Warner RR 7137343920102066447110.1097/MPA.0b 013e 3181 ebaffd · doi ↗ · pubmed ↗
- 4Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes Cancer Hallet J Law CH Cukier M Saskin R Liu N Singh S 58959712120152531276510.1002/cncr.29099 · doi ↗ · pubmed ↗
- 5Incidence of gastroenteropancreatic neuroendocrine tumours: a systematic review of the literature Endocr Relat Cancer Fraenkel M Kim M Faggiano A de Herder WW Valk GD 06321201410.1530/ERC-13-012524322304 · doi ↗ · pubmed ↗
- 6Chromogranin A: is it a useful marker of neuroendocrine tumors?J Clin Oncol Campana D Nori F Piscitelli L Morselli-Labate AM Pezzilli R Corinaldesi R Tomassetti P 196719732520071751380210.1200/JCO.2006.10.1535 · doi ↗ · pubmed ↗
- 7Appropriate use criteria for somatostatin receptor PET imaging in neuroendocrine tumors J Nucl Med Hope TA Bergsland EK Bozkurt MF 66745920182902598210.2967/jnumed.117.202275 PMC 6910630 · doi ↗ · pubmed ↗
- 8The 2019 WHO classification of tumours of the digestive system Histopathology Nagtegaal ID Odze RD Klimstra D 1821887620203143351510.1111/his.13975 PMC 7003895 · doi ↗ · pubmed ↗