Recognizing Haemophagocytic Lymphohistiocytosis in an HIV Patient With Disseminated Tuberculosis: Not Every Fever is Sepsis
Kawser Ahmed, Ashfaq Hamid

TL;DR
This paper presents a case of a rare immune condition in an HIV patient with tuberculosis, highlighting the importance of recognizing it to avoid misdiagnosis as sepsis.
Contribution
The paper emphasizes the diagnostic challenges of HLH in patients with HIV and TB, offering insights into clinical management.
Findings
The patient had a high HScore (272) despite no evidence of haemophagocytosis in the bone marrow.
Bronchial washing confirmed the presence of Mycobacterium tuberculosis.
Immunosuppressive therapy combined with antiretroviral and anti-tubercular treatment led to clinical improvement.
Abstract
Haemophagocytic lymphohistiocytosis (HLH) is a rare but potentially life-threatening condition that causes hyperinflammation, extensive tissue destruction, multi-organ failure (MOF) and death due to uncontrolled activation and proliferation of T cells and macrophages. HLH can be primary (genetic) and secondary (acquired). Primary HLH occurs due to genetic mutations, and secondary HLH is often triggered by autoimmune diseases, infections (commonly viral, bacterial and parasites) and malignancies, such as lymphoma. We report a case of secondary HLH in a 43-year-old man with an untreated human immunodeficiency virus (HIV) infection and newly diagnosed disseminated tuberculosis who was admitted to the acute medicine ward with some non-specific gastrointestinal symptoms. Nevertheless, he was treated with guideline-directed antibiotics, but his condition failed to improve with persistent…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
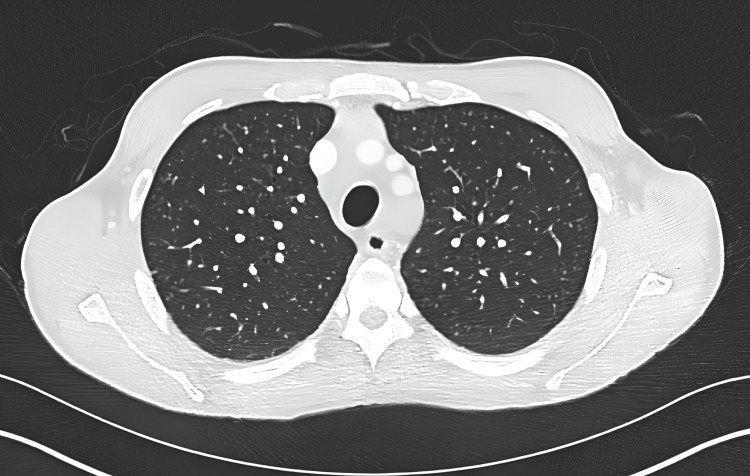 Figure 1
Figure 1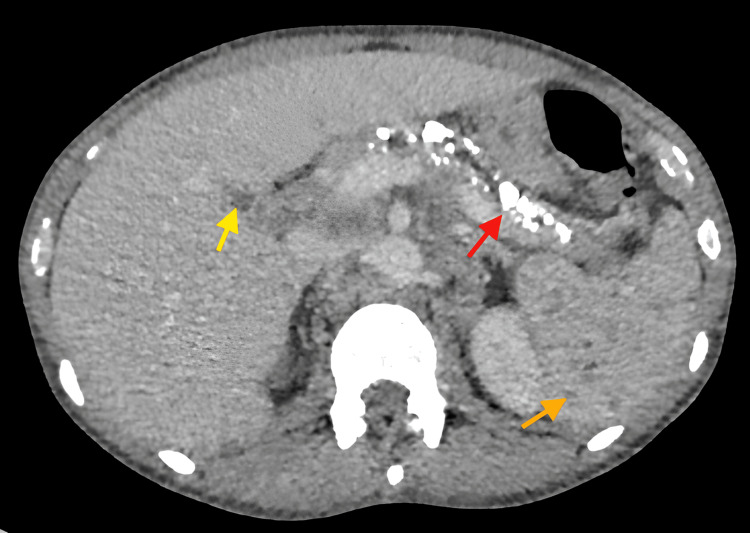 Figure 2
Figure 2| Test | Result | Normal range |
| Hb | 71 g/L | 120-150 g/L |
| Hct | 23% | 40%-50% |
| WCC | 7.3 × 10⁹/L | 4-10 × 10⁹/L |
| Neutrophil | 6.87 × 10⁹/L | 1.5-6.5 × 10⁹/L |
| Plt | 139 × 10⁹/L | 150-400 × 10⁹/L |
| CRP | 275 mg/L | <5 mg/L |
| Na | 130 mmol/L | 135-145 mmol/L |
| K | 3.1 mmol/L | 3.5-5.1 mmol/L |
| Urea | 4.5 mg/dL | 7-20 mg/dL |
| Creatinine | 65 mg/dL | 59-101 mg/dL |
| eGFR | >90 mL/min | >60 mL/min |
| Total protein | 64 g/L | 63-82 g/L |
| Albumin | 29 g/L | 35-50 g/L |
| cCa²⁺ | 2.32 mmol/L | 2.25-2.65 mmol/L |
| Bilirubin | 17 μmol/L | 3-22 μmol/L |
| ALP | 66 IU/L | 38-126 IU/L |
| ALT | 94 IU/L | 9-52 IU/L |
| APTT | 36 seconds | 22-30 seconds |
| INR | 1.1 | 0.8-1.2 |
| TSAT | 8% | 20%-50% |
| Iron | 2 μmol/L | 14-32 μmol/L |
| TIBC | 26 μmol/L | 44-71 μmol/L |
| LDH | 725 IU/L | 13-225 IU/L |
| CXR | Unremarkable | - |
| Test | Result | Normal range |
| Hb | 65 g/L | 120-150 g/L |
| Hct | 22% | 40%-50% |
| WCC | 1.1 × 10⁹/L | 4-10 × 10⁹/L |
| Neutrophil | 0.97 × 10⁹/L | 1.5-6.5 × 10⁹/L |
| Plt | 61 × 10⁹/L | 150-400 × 10⁹/L |
| CRP | 326 mg/L | <5 mg/L |
| Na⁺ | 134 mmol/L | 135-145 mmol/L |
| K⁺ | 3.2 mmol/L | 3.5-5.1 mmol/L |
| Urea | 3.7 mg/dL | 7-20 mg/dL |
| Creatinine | 52 mg/dL | 59-101 mg/dL |
| eGFR | >90 mL/min | >60 mL/min |
| Ferritin | 18,283 μg/L | 30-400 μg/L |
| Total protein | 53 g/L | 63-82 g/L |
| Albumin | 21 g/L | 35-50 g/L |
| cCa²⁺ | 2.32 mmol/L | 2.25-2.65 mmol/L |
| Total serum bilirubin | 14 μmol/L | 3-22 μmol/L |
| ALP | 145 IU/L | 38-126 IU/L |
| ALT | 66 IU/L | 9-52 IU/L |
| AST | 222 IU/L | 5-40 IU/L |
| Triglycerides | 2.7 mmol/L | 0.0-2.3 mmol/L |
| APTT | 41 seconds | 22-30 seconds |
| INR | 1.2 | 0.8-1.2 |
| Fibrinogen | 5.1 g/L | 1.8-5.0 g/L |
| LDH | 1,131 IU/L | 13-225 IU/L |
| Traits | Positive features | Score |
| Known immunosuppression | Yes (HIV) | 18 |
| Temperature | >39.4°C | 49 |
| Organomegaly | Yes (splenomegaly) | 23 |
| Cytopaenia | 3 lineages | 34 |
| Ferritin | >6,000 μg/L | 50 |
| Triglyceride | 2.7 mmol/L | 44 |
| Fibrinogen | >250 mg/dL | 0 |
| AST | >30 IU/L | 19 |
| Haemophagocytosis on BN | No | 0 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Parvovirus B19 Infection Studies · Immune Cell Function and Interaction
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a systemic hyperinflammatory disorder caused by a dysregulated immune system, which leads to cytokine storm, widespread tissue injury and multi-organ failure, and is associated with a significantly high mortality rate (ranging from 26.5% to 748% in reported series) if left untreated [1]. Primary HLH is more common in children, and the defective genes are UNC13D, PRF1, STX11 and STXBP2 [2]. Secondary HLH is primarily found in adults and is typically associated with external triggers such as autoimmune disorders, infections, cancers, immunodeficiency states and medications [3]. Regardless of the aetiology, the pathophysiology culminates in a 'cytokine storm' due to the uncontrolled activation and proliferation of cytotoxic T cells and macrophages [4]. The diagnosis of HLH, especially in the acute medicine ward, is truly challenging due to its similar presentation to severe sepsis, systemic inflammatory response syndrome (SIRS) or cytokine storm [5]. The typical presentation of HLH is persistent high fever, lymphadenopathy, organomegaly, cytopaenias, hyperferritinaemia and transaminitis [6]. The hallmark of HLH is the presence of haemophagocytes in the bone marrow; however, this finding is time-consuming to detect and may be negative in the early stages of the disease process [7]. Therefore, diagnostic tools such as the HLH-2004 criteria, as well as the Haemophagocytic Lymphohistiocytosis Diagnostic Score (HScore), are the initial assessment methods to pave the diagnostic pathway [8].
Secondary HLH, especially infections triggered, is particularly important for immunosuppressed patients. HLH due to human immunodeficiency virus (HIV) is commonly associated with opportunistic infections such as tuberculosis (TB), Epstein-Barr virus (EBV), cytomegalovirus (CMV) and histoplasmosis [9]. Disseminated TB is a recognized trigger for secondary HLH, particularly in advanced HIV infection characterized by low CD4 counts and high viral loads [10]. It should be noted that immune reconstitution inflammatory syndrome (IRIS) is usually diagnosed after the initiation of antiretroviral therapy (ART), whereas the patient in this case report had untreated HIV [11]. Therefore, this case report highlights the importance of early recognition and treatment of HLH in immunocompromised patients presenting with systemic inflammatory features unresponsive to standard therapy. The treatment approach of primary HLH, which often involves allogenic bone marrow transplantation combined with steroids and chemotherapy, differs significantly from the management of secondary HLH [12].
Case presentation
A 43-year-old male patient presented to the emergency department with multiple episodes of diarrhoea, abdominal pain and weight loss for one week in conjunction with malaise. He denied any associated vomiting, haematemesis, PR bleeding, faecal urgency, headache, chest pain or short of breath (SOB).
His past medical illness was significant for untreated HIV, diagnosed in 2023, and pulmonary embolism in 2024. He was not on any regular medications and had no known allergies.
On initial examinations, his National Early Warning Score (NEWS) was 2. Observations revealed tachycardia (heart rate (HR): 112 bpm, regular), blood pressure of 120/86 mmHg, temperature of >36.1°C, respiratory rate (RR) of 18 breaths/minute and saturations of 96% on room air. He opened his bowel six times with type 5 stool as per the Bristol stool chart over the past 24 hours. The patient was alert and did not require any supplemental oxygen therapy. Abdominal examinations showed a soft but tender abdomen, which limited further evaluation. Additionally, there were a few small, non-tender anterior cervical lymphadenopathies. His other systemic reviews were unremarkable. Initial venous blood gas (VBG) showed pH of 7.8, haemoglobin (Hb) of 78 g/L, hypokalaemia (potassium (K^+^): 2.9 mmol/L), hyponatraemia (sodium (Na^+^): 129 mmol/L) and lactate of 1.3 mmol/L. However, the routine bloods showed the following findings (Table 1), where some discrepancies were noted due to differences in values between the VBG machine and laboratory values.
Based on his presentation, infectious diarrhoea was suspected initially, and he was treated with intravenous (IV) empiric antibiotic and IV fluid, and faecal testings were requested. However, stool cultures, Clostridium difficile toxin (CDT) and CMV PCR returned negative. Infectious mononucleosis was also excluded with a negative Monospot test.
His initial differentials were acute diarrhoea, probably due to infectious causes, such as CMV, HIV wasting syndrome and infectious mononucleosis. His stool test was negative, along with a negative Monospot test. However, he was initially managed with IV antibiotics, IV fluids and maintaining stool chart.
Despite the supportive treatment as per Trust protocol, his condition deteriorated over the following days. He was experiencing persistently high-grade fever (>39.8°C), violent rigors confused as seizure-like activities, new-onset confusion and progressive breathlessness. His NEWS remained persistently high. Therefore, a computed tomography (CT) of the chest, abdomen and pelvis was performed, where positive findings are noted in Figure 1 and Figure 2.
CT of the thorax (axial view) showing multiple tiny lung nodules bilaterally with the differential of calcification and miliary TBCT: computed tomography, TB: tuberculosis
CT of the abdomen (axial view) showing multiple tiny hypoenhancing splenic nodules, extensive pancreatic calcification and one hypoenhancing liver noduleGolden arrow: spleen, red arrow: pancreas, yellow arrow: liverCT: computed tomography
Serological testing is positive for both anti-HIV-1 and p24, with a high viral load (1.70 × 106 copies/mL). Epstein-Barr virus (EBV) and parvovirus B19 serology were negative. CMV PCR and C. difficile PCR remained undetectable. However, bronchoscopy followed by bronchial washing confirmed Mycobacterium tuberculosis (TB).
Despite broad-spectrum antibiotic, antiviral and anti-TB regimens, his clinical conditions deteriorated by day 10 of admission (Table 2). He exhibited continuous high-grade fever (40.6°C), tachycardia (HR: 128 bpm, regular), low pulse pressure, hypotension and tachypnoea (RR: 32 breaths/minute), and required supplemental oxygen. He had a pseudoseizure due to violent rigor. Repeat examinations revealed cervical lymphadenopathy and splenomegaly. He underwent peri-arrest call, and repeat CT and CXR revealed no new changes. At this point, HLH was clinically suspected due to the constellation of features that are characteristic of this syndrome. These findings prompted involvement of a multidisciplinary team (MDT) comprising acute medicine, rheumatology, haematology and infectious disease specialists. The MDT outcome was to follow the British Society of Rheumatology HLH pathway, and the calculated Haemophagocytic Lymphohistiocytosis Diagnostic Score (HScore) is shown in Table 3.
The calculated HScore was 272, consistent with a high probability of HLH. The MDT mutually agreed to label it as HLH in the context of untreated HIV infection and disseminated TB. Therefore, he was promptly treated with IV pulse methyl prednisolone 1 gm for three consecutive days along with antiretroviral and anti-TB regimen and supportive care. After three days of pulse methylprednisolone, his condition improved both clinically and biochemically, which also supported the diagnosis of HLH. The next day, he was transferred to a tertiary HLH centre for further management and was followed up there.
Discussion
Haemophagocytic lymphohistiocytosis is a severe, life-threatening hyperinflammatory syndrome caused by excessive immune activation, most commonly involving T lymphocytes and macrophages [13]. This immune dysregulation results in an uncontrolled cytokine storm, leading to multiorgan failure and death if not recognized and treated early [14]. HLH is broadly classified into primary (genetic) and secondary (acquired) types [15]. While primary HLH typically manifests in children due to mutations in genes such as PRF1, UNC13D, STX11 and STXBP2, secondary HLH is more common in adults and is usually triggered by infections, cancers, autoimmune disorders or immunosuppressive therapy [16].
We report a case of a 43-year-old man with untreated HIV infection and disseminated tuberculosis (TB) presenting with features highly suggestive of HLH. The constellation of signs and laboratory findings, including persistent high fever, trilineage cytopaenias, splenomegaly, marked hyperferritinaemia, hypertriglyceridaemia and transaminitis, along with an exceptionally high HScore of 272, strongly suggested the diagnosis of HLH. Subsequently, the MDT confirmed the diagnosis based on the current diagnostic guideline, although the bone marrow was negative for haemophagocytosis.
HLH in HIV-positive patients poses a significant diagnostic challenge due to overlapping features with opportunistic infections and immune reconstitution inflammatory syndrome (IRIS) [9]. Although HIV does not directly cause HLH, the opportunistic infections associated with HIV, such as EBV, CMV, histoplasmosis and TB, are known to cause secondary/acquired HLH [10]. In our patient, TB was the inciting factor on the background of HIV, as we excluded the other opportunistic infections based on the blood test, history and examinations.
TB-associated HLH is becoming more widely acknowledged, especially in immunocompromised individuals, in the literature. A recent systematic review highlighted that TB is one of the most prevalent infectious causes of HLH in HIV-positive patients [17]. As seen in our patient, disseminated TB is frequently linked to HLH and manifests as fever, weight loss, cytopaenias and raised inflammatory markers.
Distinguishing HLH from severe sepsis or systemic inflammatory response syndrome (SIRS) is a major clinical challenge [18]. Since our patient was not on antiretroviral therapy (ART) when symptoms began, immune reconstitution inflammatory syndrome (IRIS) was excluded as an initial diagnosis. Furthermore, the presence of extreme hyperferritinaemia (18,000ug/L in this case), in conjunction with progressive cytopaenias and hepatic dysfunction, strongly favoured HLH over uncomplicated sepsis or disseminated TB. Consideration of HLH is prudent by these clinical and biochemical indicators when the patient was not improving with the conventional antimicrobial treatment.
The HLH-2004 diagnostic criteria or the more recent revised diagnostic guidelines are used to diagnose HLH. These criteria need a molecular diagnosis or the fulfillment of more than five out of eight clinical and laboratory parameters [19]. Our patient satisfied the requirements of fever, splenomegaly, cytopaenias in three lineages, hyperferritinaemia and hypertriglyceridaemia. Despite being the hallmark of the bone marrow or tissue biopsies, haemophagocytosis is neither sensitive nor specific, although it might be absent in the early stage of the disease [20]. Therefore, physicians should not exclusively rely on the biopsy result, as without the immediate treatment, patients might deteriorate quickly [21]. Hence, the HScore has been established with the combinations of clinical and laboratory markers to determine the probability of adult HLH. A score more than 250 is associated with a >99% chance of adult HLH [22]. Our patient's HScore of 272 provided strong support for the diagnosis of HLH, facilitating the urgent therapeutic action.
Two main strategies are needed to manage secondary HLH: immunosuppression to control hyperinflammation and specific treatment of the secondary causes [23]. The equilibrium is more delicate in infection-triggered secondary HLH. The mainstay of treatment of HLH is high-dose steroids [24]. Our patient had a dramatic improvement in both clinical and biochemical markers after starting the intravenous pulse methylprednisolone (1 gm daily for three subsequent days). At the same time, an anti-TB regimen was started. The requirement of high-dose steroids in the middle of an active infection makes the management of TB in HLH more challenging [25]. According to recent research, steroids and anti-TB regimens work better together to increase the survival rates in TB-associated HLH [26]. Antiretroviral therapy was also introduced, although its timing was carefully considered to reduce the risk of IRIS.
Etoposide, an anti-cancer drug, has an established role in paediatric HLH and in severe or refractory adult HLH [27]. However, our patient did not receive this treatment. Referral to a tertiary HLH centre, along with an MDT approach, is key to managing HLH, which was also contemplated in our patient [21]. The complexities of HLH treatment demand such collaboration, which is crucial.
Recent research highlights the increasing prevalence of HLH among immunocompromised individuals, especially those with co-infection of HIV and TB [17]. A recent review article stressed that HLH should be considered in patients who presented with fever, cytopaenia and raised ferritin, even if the alternative diagnosis of sepsis and tuberculosis is suspected initially [28]. Data from the HLH registry suggests that the outcome is fatal if the diagnosis and treatment are delayed [29]. Therefore, it is crucial to use the diagnostic algorithm rather than waiting for a bone marrow biopsy to start immunosuppressant and the treatment of inciting factors [30].
Conclusions
This case underscores the critical importance of maintaining a high index of suspicion for secondary HLH in immunocompromised patients, specifically those with co-existing untreated HIV and disseminated TB, who present with persistent systemic inflammation and multi-organ dysfunction. Further research is warranted to develop simplified diagnostic pathways for HLH in low-resource settings. This case reinforces the need for institutional protocols to streamline referral to specialized centres. Greater awareness and training on HLH recognition among acute care teams may facilitate earlier intervention and improve survival.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Haemophagocytic lymphohistiocytosis (HLH) in an adult following treatment with trimethoprim/sulfamethoxazole BMJ Case Rep Zimilover A Huml I 15202210.1136/bcr-2022-251255 PMC 975617536517076 · doi ↗ · pubmed ↗
- 2Haemophagocytic lymphohistiocytosis: an elusive syndrome Clin Med (Lond) Thomas W Veer MV Besser M 4324361620162769780410.7861/clinmedicine.16-5-432PMC 6297301 · doi ↗ · pubmed ↗
- 3Case report: primary hemophagocytic syndrome triggered by dengue infection ID Cases Devi K Ali N 026202110.1016/j.idcr.2021.e 01275 PMC 841388334504769 · doi ↗ · pubmed ↗
- 4Pathogenesis of hemophagocytic lymphohistiocytosis/macrophage activation syndrome : a case report and review of the literature Int J Mol Sci Gioia C Paroli M Izzo R Di Sanzo L Rossi E Pignatelli P Accapezzato D 25202410.3390/ijms 25115921 PMC 1117313338892108 · doi ↗ · pubmed ↗
- 5Diagnostic challenges of hemophagocytic lymphohistiocytosis Clin Lymphoma Myeloma Leuk Otrock ZK Daver N Kantarjian HM Eby CS 01017 S 201710.1016/j.clml.2017.02.01728760295 · doi ↗ · pubmed ↗
- 6Hemophagocytic lymphohistiocytosis: epidemiological, clinical and biological profile Turk J Med Sci Zahir H Belkhir J Mouhib H Ait Ameur M Chakour M 133213354920193164843810.3906/sag-1812-65PMC 7018207 · doi ↗ · pubmed ↗
- 7Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability Am J Clin Pathol Ho C Yao X Tian L Li FY Podoltsev N Xu ML 627114120142434373810.1309/AJCPMD 5TJEFOOVBW · doi ↗ · pubmed ↗
- 8Multicenter validation of secondary hemophagocytic lymphohistiocytosis diagnostic criteria J Intern Med Lachmann G Heeren P Schuster FS 31232729720253986885210.1111/joim.20065 PMC 11846073 · doi ↗ · pubmed ↗