Lacrimispora sanguinis sp. nov., isolated from human blood
Hui-Jin Yu, Yun Young Cho, Jayoung Paek, Minhee Kang, Mi Young Ahn, Heejung Kim, Jung-Hyun Byun, Tae Yeul Kim, Hee Jae Huh, Lu Bai, Young-Hyo Chang

TL;DR
Scientists discovered a new species of bacteria, Lacrimispora sanguinis, isolated from human blood, and found it is closely related to other Lacrimispora strains.
Contribution
The proposal of a new bacterial species, Lacrimispora sanguinis, based on genomic, chemotaxonomic, and phenotypic analyses.
Findings
Strain HJ-01T forms a distinct cluster adjacent to L. celerecrescens strains in phylogenomic analysis.
The strain shares high resistance to clindamycin due to the cfr(C) gene.
The new species is characterized by specific fatty acids and peptidoglycan composition.
Abstract
A rod-shaped, obligate anaerobic, Gram-stain-positive bacterium isolated from the human blood was designated as the strain HJ-01T. Analysis of the 16S rRNA gene sequence revealed that the strain HJ-01T belonged to the genus Lacrimispora, and was most closely related to L. celerecrescens strains DSM 105336 and MCM B-936, with both 99.3% similarity. The average nucleotide identity values between the strain and the most closely related type strains ranged from 75.3% to 91.4%, while the values between the strain and the two non-type strains of L. celerecrescens, DSM 105336 and MCM B-936, were 98.8% to 98.9%. The digital DNA-DNA hybridization values between the strain and the most closely related type strains ranged from 19.8% to 44.5%, whereas the values between the strain and L. celerecrescens strains DSM 105336 and MCM B-936 were 89.7% to 91.6%. The phylogenomic analysis revealed that the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
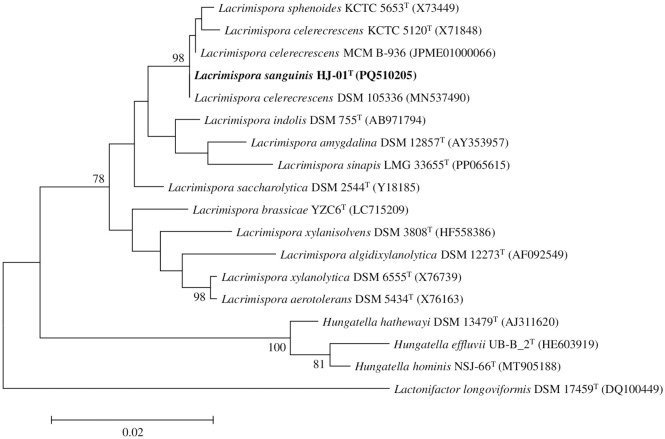 Fig 1
Fig 1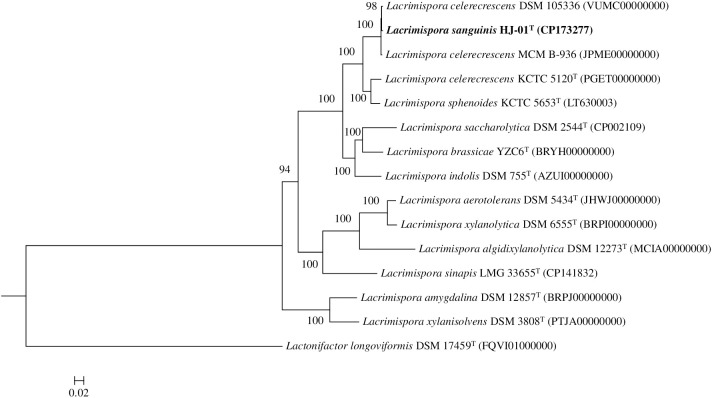 Fig 2
Fig 2- —http://dx.doi.org/10.13039/501100003710Korea Health Industry Development Institute
- —http://dx.doi.org/10.13039/501100003715Korea Research Institute of Bioscience and Biotechnology
- —Samsung Medical Center
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Bacterial Identification and Susceptibility Testing · Infective Endocarditis Diagnosis and Management
Introduction
The Clostridium sphenoides group has recently been reclassified as the genus Lacrimispora within the family Lachnospiraceae [1]. This genus currently comprises 11 species with validly published names [2], with Lacrimispora sphenoides serving as the type species, and the most recent addition being Lacrimispora sinapis, isolated from pickled potherb mustard [3]. Members of the genus Lacrimispora are generally Gram-positive, spore-forming, rod-shaped, and anaerobic bacteria [1]. They are commonly isolated from environmental sources such as soil, plants, animal, and human sources, with some occasionally implicated in human infections. For instance, L. sphenoides has been reported to cause gastroenteritis, osteomyelitis, peritonitis, and bloodstream infections [4–8], while Lacrimispora celerecrescens has been associated with osteomyelitis [9,10].
Microorganisms play vital roles in all ecosystems, yet their rapid and accurate characterization remains a major challenge. The matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) has become a powerful microbial classification technique due to its speed, affordability, and ease of use, making it exceptionally valuable for detecting bacteria in clinical, environmental, and microbiome diversity research [11]. Previously, we isolated a strain HJ-01^T^ from human blood and identified it within the Lacrimispora genus by MALDI-TOF MS. The aim of the present study was to determine the taxonomic status of this strain using a polyphasic approach.
Materials and methods
Ethics statement
This study was reviewed and approved by the Institutional Review Board of Seoul Medical Center (approval no. SEOUL 2024-08-007). The requirement for informed consent was waived as the study involved only bacterial strains obtained through routine diagnostic testing and public culture collections, with all patient data fully anonymized. Patient data were retrospectively accessed for research purposes from 01/10/2024 to 30/11/2024.
Strain isolation and patient history
Strain HJ-01^T^ was isolated from the blood of an 81-year-old woman with hypertension and no other significant comorbidities. Following intravenous infusion of an albumin solution not authorized for clinical use by the Korean Ministry of Food and Drug Safety at the time, she was presented to the emergency room with signs of sepsis. Her initial vital signs were: body temperature of 39.6°C, blood pressure of 92/47 mm Hg, pulse rate of 108 beats per minute, respiratory rate of 22 breaths per minute, and oxygen saturation of 92% while breathing ambient air. Two sets of blood cultures were obtained, and empirical antibiotic therapy with meropenem was initiated. After overnight incubation, Gram-positive bacilli were detected in the anaerobic blood culture bottles, prompting the addition of teicoplanin. The anerobic, Gram-positive bacilli were subcultured on Brucella blood agar supplemented with 5% (v/v) sheep blood and incubated anaerobically at 36°C for 3 days. Strain HJ-01^T^ was identified using MALDI-TOF MS (MALDI Biotyper; Bruker, Bremen, Germany) and preserved in skim milk (BD Difco Skim Milk) at −80°C until further use. Upon availability of the identification result, teicoplanin was discontinued, while meropenem was continued, resulting in a favorable clinical response. Subsequent blood cultures were negative, and the patient recovered fully, being discharged on day 15 without complications.
Phylogenetic analysis
16S rRNA gene amplification was performed using primers previously reported by Bai et al. [12]. The 16S rRNA gene sequence of the strain was aligned with reference sequences of published prokaryotic type strains retrieved from the GenBank and EzBioCloud databases (https://www.ezbiocloud.net/) [13]. The phylogenetic trees were generated from unambiguous alignments, with alignment conducted with PHYLIP and phylogenetic analyses carried out using jPHYDIT [14]. Whole-genome and core gene phylogenies were constructed using FastME (GBDP, distance formula d5) and UBCG2, respectively, with 100 and 1,000 bootstrap replicates. Lactonifactor longoviformis DSM 17459^T^ was designated as the outgroup, and branch lengths were scaled to 0.20 substitutions per site [15,16]. Maximum-likelihood (ML), neighbor-joining (NJ), and minimum-evolution (ME) algorithms were used to reconstruct phylogenetic trees [17]. Reliability of the phylogenetic tree was assessed by bootstrap analysis with 1000 resamplings [18]. Lactonifactor longoviformis DSM 17459^T^ was used as the outgroup.
Genomic analysis
Genomic DNA of strain HJ-01^T^ was extracted using the Wizard HMW DNA Extraction Kit following the manufacturer’s protocol. Library preparation utilized the SMRTbell protocol, with size selection (7–12 kb cutoff) performed using the Megaruptor 3 (Diagenode). Sequencing was conducted on the PacBio Revio platform (Pacific Biosciences) by Phyzen Genomics Institute (Seongnam, Republic of Korea). De novo assembly of high-fidelity (HiFi) reads was performed with Flye (v2.9.4), and assembly validation was achieved by aligning HiFi reads to assembled sequences using Pbmm2 (v1.14.0). Genome annotation was carried out using Prokka (v1.14.6). The genome map visualization and genome comparison were generated with the proksee server [19]. CheckM was used to evaluate the completeness of the genome [20]. Average nucleotide identity (ANI) values were determined using OrthoANIu [21]. Digital DNA-DNA hybridization (dDDH) values were calculated using the formula 2 of the Genome-to-Genome Distance Calculator (GGDC, version 3.0; http://ggdc.dsmz.de/ggdc.php#) [22]. In addition, a multi-locus species tree based on 100 conserved core genes, derived from the whole-genome sequences of the strain and its closely related strains, was generated using the IQ-TREE maximum likelihood method implemented in autoMLST (https://automlst.ziemertlab.com) [23]. Comprehensive Antibiotic Resistance Database (CARD) was utilized to detect antibiotic resistance genes [24]. PathogenFinder (v1.1) and VirulenceFinder (v3.0.2) were used to identify pathogenicity and virulence genes [25]. Whole-genome sequences and their corresponding GenBank accession numbers, obtained from NCBI (https://www.ncbi.nlm.nih.gov/genome), were used for ANI, dDDH, and phylogenomic analyses.
Phenotypic and chemotaxonomic analyses
The strain and its related strains were cultivated on Brucella blood plates at 36°C for 3 days prior to biochemical, physiological, and morphological characterization. For aerotolerance test, the strain was further incubated in fluid thioglycollate medium at 36°C for 3 days [26]. Cells motility was examined in semi-solid Brucella serum medium (0.3% w/v). Flagellation and cell morphology were observed using a transmission electron microscope (CM20, Philips, Netherlands) and a phase-contrast microscope (E600, Nikon, Japan). Gram-staining was carried out using a commercial Gram-staining kit (BioMérieux, France) according to the manufacturer’s instructions. Following cultivation on Brucella blood plates at 36°C for 3 days, spore formation was assessed by staining with 5% malachite green and counterstaining with safranin [27]. Gram-staining and spore formation were observed using an upright microscope (ECLIPSE Ci, Nikon, Japan). The Brucella blood medium was employed to assess tolerance to pH, temperature, and salinity. pH tolerance was tested over a range of 5.0 to 12.0 in 1-unit increments, salinity tolerance was evaluated using NaCl concentration of up to 5% (w/v), in 0.5% increments and growth temperature was determined from 4 to 60°C in 5–10°C increments [28]. Additional biochemical phenotypic characterization was preformed using API 20A and 32A kits (BioMérieux, France).
Fermentation end products were analyzed by liquid chromatography (Ultimate 3000, Thermo Dionex, USA) using an Aminex 87H column (300 × 10 mm, Bio-Rad, USA) with0.01N H_2_SO_4_ as the mobile phase at flow rate of 0.5 ml/min. Detection was performed at 210 nm using a RefractoMAX520 detector (Japan) [29]. Whole-cell fatty acid composition was analyzed after culturing the strain and reference strains on Brucella blood plates at 36°C for 3 days, followed by cell harvesting. Fatty acids profiles were determined by gas chromatography using the MOORE6 Library (version 6.0) of the MIDI/Hewlett-Packard Microbial Identification System [30]. Peptidoglycan composition was examined according to the method outlined by Schumann [31].
Antimicrobial susceptibility testing was conducted using the Etest (bioMérieux, Marcy-l’Étoile, France) following the manufacturer’s instructions against penicillin, ampicillin, amoxicillin-clavulanate, piperacillin-tazobactam, clindamycin, ertapenem, imipenem, and metronidazole. Minimum inhibitory concentrations (MICs) were interpreted according to the breakpoints outlined in the Clinical and Laboratory Standards Institute M100-Ed34 document [32].
Results and discussion
Phylogenetic analysis
The 1,461 bp 16S rRNA gene sequence of the strain HJ-01^T^ was used for phylogenetic analysis. The 16S rRNA similarities between the strain and the most closely related strains, L. celerecrescens DSM 105336 and L. celerecrescens MCM B-936, were both 99.3% (S1 Table). The ML tree placed the strain within a cluster of the genus Lacrimispora (Fig 1) forming a distinct branch adjacent to L. celerecrescens and L. sphenoides. Phylogenetic analysis with the use of the ML method demonstrated that the strain belongs to the genus Lacrimispora. Similar results were observed with the NJ (S1 Fig) and ME trees (S2 Fig).
Phylogenetic consensus tree based on 16S rRNA gene sequences, reconstructed with the maximum-likelihood (ML) method, indicating the taxonomic positions of strain HJ-01T and its close relatives.Bootstrap values (≥70%) based on 1,000 replicates are shown at branch nodes. Lactonifactor longoviformis DSM 17459T was used as an outgroup. Bar, 0.02 substitutions per nucleotide.
Genomic analysis
Overall, 921,496,507 bp of the strain HJ-01^T^ (170 × coverage) was read. The genome assembly of the strain HJ-01^T^ consisted of a single contig of 5,411,172 bp, with an N50 of 5,411,172 bp. The sequenced genome satisfied the minimal standards for quality [34]. The genome completeness of the strain was 98.1%, with 2.3% contamination. The total genome length of the strain HJ-01^T^ was 5,411,172 bp, with a G + C content of 43.2 mol% (Table 1). The G + C content of the strain was comparable to that of other species within Lacrimispora. The assembled genome of strain HJ-01^T^ contained 5,055 genes, including 4,928 protein-coding sequences (CDS), 18 rRNAs, and 69 tRNAs (S5 Fig).
Table 1: Genomic characteristics of strain HJ-01Tand closely related species.
The genome comparison demonstrated that the strain was most closely related to L. celerecrescens DSM 105336 and L. celerecrescens MCM B-936 (S3-S6 Fig). The phylogenomic tree revealed that the strain HJ-01^T^ formed a distinct clade with L. celerecrescens strains DSM 105336 and MCM B-936. This clade was positioned adjacent to L. celerecrescens KCTC 5120^T^ and L. sphenoides KCTC 5653^T^ (Fig 2). The ANI values between the strain HJ-01^T^ and the most closely related type strains ranged from 75.3% to 91.4%, while the values between the strain and the two non-type strains of L. celerecrescens, DSM 105336 and MCM B-936, were 98.9% and 98.8%, respectively. The dDDH values between the strain and the most closely related type strains ranged from 19.8% to 44.5%, whereas the values between the strain and L. celerecrescens strains DSM 105336 and MCM B-936 were 91.6% and 89.7%, respectively (Table 2). The ANI and dDDH values indicated that the values between the strain and the related type strains within the genus Lacrimispora were below the bacterial species delineation thresholds [33]. Moreover, the strain HJ-01^T^ was most closely related to the L. celerecrescens strains DSM 105336 and MCM B-936. These results indicate that the strain represents a new species within the genus Lacrimispora, with the likelihood that L. celerecrescens DSM 105336 and L. celerecrescens MCM B-936 also belongs to this novel species.
Table 2: Average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) values (%) between strain HJ-01T and the close relatives of Lacrimispora.
Molecular phylogenetic analysis based on genome sequences of strain HJ-01T and its close relatives using autoMLST with default parameters (>100 core genes).Bootstrap values (≥70%) based on 1,000 replicates are shown at branch nodes. Lactonifactor longoviformis DSM 17459T was served as an outgroup. Bar, 0.02 substitutions per nucleotide position.
CARD analysis identified the cfr(C) gene in the genomes of the strain HJ-01^T^ and related strains, strongly associated with high clindamycin resistance. The cfr gene encodes a methyltransferase that modifies the C8 position of A2503 in 23S rRNA, conferring resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A (PhLOPSA phenotype). Since its initial discovery in Staphylococcus sciuri in 2000, five cfr variants (cfr, cfr(B), cfr(C), cfr(D), and cfr(E)) have been described [34]. The cfr(C) gene has been reported in Campylobacter coli, Clostridioides difficile, Clostridium perfringens, and Bacteroides fragilis, typically located on plasmids or chromosomes associated with transposable elements [34–37]. The identification of cfr(C) in the Lacrimispora genus is novel, representing the first report of this gene in the genus. This gene was located on the chromosome within a 3,378-bp transposed unit, flanked by two mismatched direct repeats (S7 Fig), suggesting at least two independent transposition events. The absence of transposase-encoding genes within 10 kb upstream or downstream of the transposed unit indicates its stabilization as a chromosomal feature.
Genome analysis using PathogenFinder and VirulenceFinder predicted strain HJ-01^T^ to be a non-human pathogen (0.227 probability) and no pathogenicity or virulence genes were detected. Interestingly, HJ-01^T^ matched 17 non-pathogenic protein families, all of which were also found in L. celerecrescens DSM 105336. These included 5 conserved hypothetical protein, 1 type II secretion system, 1 nucleotideyl transferase, 1 transcriptional regulator, 1 CdaR, 1 binding-protein-dependent transport systems inner membrane component, 1 ribosomal protein L17, 1 stage III sporulation protein AD, 1 protein of unknown function (DUF77), 1 ribosomal protein S6, 1 DNA-directed RNA polymerase omega subunit, 1 phosphotransferase system, 1 phosphocarrier protein HPr, and 1 protein of unknown function (DUF1540). The clinical outcomes further supported the predicted non-pathogenicity of the strain, as the patient recovered fully following appropriate antibiotic treatments. Although strain HJ-01^T^ appears to be non-pathogenic, its isolation from a clinical sample suggests clinical relevance, particularly in the context of polymicrobial communities or opportunistic colonization in compromised hosts. Similar observations have been reported for L. celerecrescens, which has been identified as an opportunistic pathogen in humans, primarily in post-traumatic or wound-related infections with low known pathogenicity and no identified virulence genes [38,39].
Phenotypic and chemotaxonomic analyses
The closely related strains L. celerecrescens DSM 105336, L. celerecrescens KCTC 5120^T^, and L. sphenoides KCTC 5653^T^ were used as reference strains. Strain HJ-01^T^ exhibited characteristic features of the genus Lacrimispora [1], being rod-shaped, spore-forming, motile, and Gram-stain-positive (S8 Fig). The spore was terminal in position and oval in shape (S9 Fig). Aerotolerance testing confirmed that the strain HJ-01^T^ is an obligate anaerobe. The strain grew at 15–37°C (optimal at 35–37°C), pH range of 6–9 (optimal at 7) and withstood salinity levels of up to 1.5% (w/v). API 20A revealed that HJ-01^T^ was ferments lactose, and raffinose, which was different from L. sphenoides KCTC 5653^T^. API 32A revealed that HJ-01^T^ and L. celerecrescens DSM 105336 were negative to α-galactosidase activity, unlike L. celerecrescens KCTC 5120^T^ and L. sphenoides KCTC 5653^T^. The strain hydrolyzed esculin but did not produce indole and urease. Detailed phenotypic characteristics of the strain and related strains are presented in Table 3.
Table 3: Distinguishing phenotypic characteristics between strain HJ-01T and closely related species.
The strain HJ-01^T^ produced acetic acid (892.7 mg/L), and formic acid (230.6 mg/L) as the major metabolic end products, similar to the related strains. The predominant whole-cell fatty acids (mean value > 10% of total fatty acids) were C_16:0_ (26.1%), and C_18:1_ cis 11 DMA (11.0%). These were also major components in L. celerecrescens DSM 105336 and L. sphenoides KCTC 5653^T^, albeit at different percentages, whereas C_18:1_ cis 11 DMA was absent among the dominant whole-cell fatty acid in L. celerecrescens KCTC 5120^T^ (Table 4). Analysis of cell wall peptidoglycan indicated that the strain and its closely related strains contained meso-diaminopimelic acid (meso-Dpm) as the diagnostic amino acid. The MIC results for strain HJ-01^T^ and the reference strains showed susceptibility to most of the tested antibiotics, including penicillin, ampicillin, amoxicillin-clavulanate, piperacillin-tazobactam, ertapenem, imipenem, and metronidazole (S2 Table), aligning with previous studies [7,8,10]. However, all the strains were resistant to clindamycin. Collectively, the phenotypic and chemotaxonomic characteristics distinguished the strain from its closely related strains.
Table 4: Cellular fatty acid profiles of strain HJ-01T and closely related species.
In conclusion, the representative peptidoglycan type of the strain HJ-01^T^ was meso-Dpm, consistent with that of related strains within the genus Lacrimispora. The end products of the fermentation were acetic acid and formic acid, and the main cellular fatty acids were C_16:0_ and C_18:1_ cis 11 DMA, which was similar to the related strains within the genus Lacrimispora. 16S rRNA phylogenetic analysis showed that the strain formed a distinct cluster within the Lacrimispora genus. The G + C content of the HJ-01^T^ genome was 43.2 mol%. The ANI values between the strain and the related species in Lacrimispora ranged from 75.3% to 91.4%, while the values between the strain and L. celerecrescens strains DSM 105336 and MCM B-936 were 98.8–98.9%. The dDDH values between the strain and the related species in Lacrimispora ranged from 19.8% to 44.5%, whereas the values between the strain and L. celerecrescens strains DSM 105336 and MCM B-936 were 89.7–91.6%. Therefore, the strain HJ-01^T^ represents a new species within the genus Lacrimispora, and the name Lacrimispora sanguinis sp. nov. is proposed. Additionally, our data suggest that L. celerecrescens DSM 105336 and MCM B-936 may also belong to Lacrimispora sanguinis sp. nov.
The strain was isolated from human blood, and while intravenous infusion may represent a possible route of entry, the infection pathway remains uncertain. As an opportunistic microorganism, host factors such as patient age may have contributed to infection; however, it cannot be confirmed that the bacterium directly caused the observed clinical symptoms.
This study underscores the significance of identifying L. sanguinis sp. nov., contributing to a broader understanding of the microbial diversity associated with a hospitalized patient. Furthermore, it highlights the value of genome-based taxonomy in delineating species boundaries and guiding future research into the ecological roles and potential clinical significance of novel anaerobic bacteria.
Description of Lacrimispora sanguinis sp. nov.
Lacrimispora sanguinis (san’gui.nis. L. n. sanguis, blood; referring to the strain isolation from human blood).
Cells are obligately anaerobic, Gram-stain-positive, spore-forming, rod-shaped, and motile by means of peritrichous flagella. Cells measure 0.5–1.0 µm in diameter, and 1.3–4.1 μm long. Colonies grown anaerobically on Brucella blood agar (5% sheep blood) at 36°C for 3 days are cream-colored. Cells grow in the range 15–37°C, at pH 6–9, with optimal growth at 35−37°C and pH 7. The species tolerance of NaCl is up to 1.5% (w/v). The end products are acetic acid and formic acid. Based on the API 20A, the strain ferments glucose, mannitol, lactose, maltose, salicin, xylose, arabinose, celiobiose, mannose, raffinose, rhamnose, and trehalose; but does not ferment saccharose, gelatine, glycerol, melezitose, and sorbitol. Based on the API 32A, the strain produces β-galactosidase, α-glucosidase, β-glucosidase, β-arabinosidase, glutamic acid, and alkaline phosphatase; but do not produce arginine dihydrolase, α-galactosidase, β-galactosidase-6-phosphate, β-glucuronidase, N-acetyl-β-glucosaminidase, D-raffinose, D-mannose, α-fucosidase, reduction of nitrates, arginine arylamidase, proline arylamidase, phenylalanine arylamidase, leucyl-glycine arylamidase, leucine arylamidase, glycine arylamidase, pyroglutamic acid arylamidase, alanine arylamidase, tyrosine arylamidase, histidine arylamidase, serine arylamidase, and glutamyl -glutamic acid arylamidase activities. Esculin is hydrolyzed, whereas indole and urease are not produced. The strain is catalase- and oxidase-negative. The predominant fatty acids are C_16:0_ and C_18:1_ cis 11 DMA. The cell-wall peptidoglycan contains meso-diaminopimelic acid as a diagnostic amino acid. The strain is susceptible to penicillin, ampicillin, amoxicillin-clavulanate, piperacillin-tazobactam, ertapenem, imipenem, and metronidazole, but resistant to clindamycin.
Strain HJ-01^T^ (KCTC 25933^T^ = JCM 37550^T^) was isolated from human blood. Its genome is 5,411,172 bp in length, with a G + C content of 43.2 mol%.
Supporting information
S1 FigPhylogenetic consensus tree based on 16S rRNA gene sequence of strain HJ-01^T^, reconstructed with the neighbor-joining (NJ), indicating the taxonomic positions of isolate and close relatives.Bootstrap values (≥70%) based on 1,000 subsets are shown at branch nodes. Lactonifactor longoviformis DSM 17459^T^ was used as an outgroup. Bar, 0.02 substitutions per nucleotide.(DOCX)
S2 FigPhylogenetic consensus tree based on 16S rRNA gene sequence of strain HJ-01^T^, reconstructed with the minimum-evolution (ME), indicating the taxonomic positions of isolate and the close relatives.Bootstrap values (≥70%) based on 1,000 subsets are shown at branch nodes. Lactonifactor longoviformis DSM 17459^T^ was used as an outgroup. Bar, 0.02 substitutions per nucleotide.(DOCX)
S3 FigPhylogenomic tree based on whole genome sequences showing the relationships between strain HJ-01^T^ and its closely related strains within the genus *Lacrimispora.*Tree inferred with FastME 2.1.6.1 [40] from GBDP distances calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values > 60% from 100 replications, with an average branch support of 98.4%. The tree was rooted at the midpoint [41].(DOCX)
S4 FigPhylogenomic tree based on core gene sequence by UBCG2 showing the relationships between strain HJ-01^T^ and its closely related strains within the genus *Lacrimispora.*Bootstrap values based on 1000 replications are listed as percentages at branch points. Bar, 0.20 substitutions per site. Lactonifactor longoviformis DSM 17459^T^ was used as an outgroup.(DOCX)
S5 FigA circular genome map of strain HJ-01ᵀ.The second and third inner circles display the G + C content and G + C skew, respectively.(DOCX)
S6 FigGenome comparison of strain HJ-01ᵀ and closely related strains within the genus *Lacrimispora.*Starting from the innermost ring, rings 1 and 2 represent the GC skew (purple/green) and GC content (black) of strain HJ-01ᵀ. Rings 3 and 4 display protein-coding genes (purple), tRNA genes (orange), tmRNA genes (green), rRNA genes (pink), and repeat regions (blue) on the forward and reverse strands of strain HJ-01ᵀ. The remaining rings show genome comparisons of L. celerecrescens DSM 105336 (ring 5), L. celerecrescens MCM B-936 (ring 6), L. sphenoides KCTC 5653ᵀ (ring 7), and L. celerecrescens KCTC 5120^T^ (ring 8) with strain HJ-01ᵀ.(DOCX)
S7 FigSchematic presentation of the vicinity of the cfr(C) gene in the chromosome of strain HJ-01ᵀ.Solid arrows indicate the positions and orientations of the open reading frames, and their colors are based on the estimated function of encoded proteins. Arrow heads indicate direct repeats.(DOCX)
S8 FigScanning electron micrograph (SEM) and transmission electron micrograph (TEM) of the strain HJ-01^T^.A, SEM image of HJ-01^T^ (bar, 5 µm); and B, TEM image of HJ-01^T^ (bar, 1 µm).(DOCX)
S9 FigTransmission electron micrograph (TEM) of the strain HJ-01^T^ endospore formation and spore.A, TEM image of HJ-01^T^ endospore (bar, 500 nm); and B, TEM image of HJ-01^T^ spore (bar, 200 nm).(DOCX)
S1 Table16S rRNA similarities between strain HJ-01^T^ and the close relatives of *Lacrimispora.*Strains: 1, HJ-01^T^; 2, L. celerecrescens DSM 105336; 3, L. celerecrescens MCM B-936; 4, L. celerecrescens KCTC 5120^T^; 5, L. sphenoides KCTC 5653^T^; 6, L. indolis DSM 755^T^; 7, L. saccharolytica DSM 2544^T^; 8, L. brassicae YZC6^T^; 9, L. amygdalina DSM 12857^T^; 10, L. xylanolytica DSM 6555^T^; 11, L. aerotolerans DSM 5434^T^; 12, L. sinapis LMG 33655^T^; 13, L. xylanisolvens DSM 3808^T^; 14, L. algidixylanolytica DSM 12273^T^.(DOCX)
S2 TableAntimicrobial susceptibility patterns of strain HJ-01^T^ and its closely related strains.Strains: 1, L. sanguinis HJ-01^T^; 2, L. celerecrescens DSM 105336; 3, L. sphenoides KCTC 5653^T^; 4, L. celerecrescens KCTC 5120^T^. All data were obtained from the current study. S, susceptible; I, intermediate; R, resistant.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Haas KN, Blanchard JL. Reclassification of the Clostridium clostridioforme and Clostridium sphenoides clades as Enterocloster gen. nov. and Lacrimispora gen. nov., including reclassification of 15 taxa. Int J Syst Evol Microbiol. 2020;70(1):23–34. doi: 10.1099/ijsem.0.003698 31782700 · doi ↗ · pubmed ↗
- 2Parte AC, Sardà Carbasse J, Meier-Kolthoff JP, Reimer LC, Göker M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int J Syst Evol Microbiol. 2020;70(11):5607–12. doi: 10.1099/ijsem.0.004332 32701423 PMC 7723251 · doi ↗ · pubmed ↗
- 3Ren Q, Wang D, Han J, Liu Z, Wu Z. Lacrimispora sinapis sp. nov., isolated from pickled potherb mustard (Brassica juncea Coss.). Int J Syst Evol Microbiol. 2025;75(2):006675. doi: 10.1099/ijsem.0.006675 39918553 PMC 11806200 · doi ↗ · pubmed ↗
- 4Felitti VJ. Primary invasion by Clostridium sphenoides in a patient with periodic neutropenia. Calif Med. 1970;113(3):76–8. 5457520 PMC 1501556 · pubmed ↗
- 5Isenberg HD, Lavine LS, Painter BG, Rubins WH, Berkman JI. Primary osteomyelitis due to an anaerobic microorganism. Am J Clin Pathol. 1975;64(3):385–8. doi: 10.1093/ajcp/64.3.385 1163490 · doi ↗ · pubmed ↗
- 6Sullivan SN, Darwish RJ, Schieven BC. Severe diarrhea due to Clostridium sphenoides: a case report. Can Med Assoc J. 1980;123(5):398. 7260781 PMC 1704784 · pubmed ↗
- 7Kelesidis T, Tsiodras S. Clostridium sphenoides bloodstream infection in man. Emerg Infect Dis. 2011;17(1):156–8. doi: 10.3201/eid 1701.101029 21192891 PMC 3204644 · doi ↗ · pubmed ↗
- 8Perkins MJ, Snesrud E, Mc Gann P, Duplessis CA. Clostridium sphenoides Chronic Osteomyelitis Diagnosed Via Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry, Conflicting With 16S r RNA Sequencing but Confirmed by Whole Genome Sequencing. Mil Med. 2017;182(1):e 1669–72. doi: 10.7205/MILMED-D-15-00535 28051992 · doi ↗ · pubmed ↗