Genomic characteristics of Lacticaseibacillus rhamnosus strains isolated from blood
Piotr Jarocki, Jan Sadurski, Martyna Siuda, Mateusz Romanowicz, Jacek Panek, Magdalena Frąc, Adam Waśko

TL;DR
This study analyzes the genomes of Lacticaseibacillus rhamnosus strains from blood, revealing their genetic diversity and dual potential for health benefits and pathogenicity.
Contribution
The study provides complete genomes of L. rhamnosus blood isolates and identifies genetic traits linked to both probiotic and pathogenic functions.
Findings
L. rhamnosus blood isolates showed genetic diversity and no close relation to the probiotic strain L. rhamnosus GG.
Certain genes linked to probiotic functions also overlap with virulence factors in pathogenic microbes.
Genomic analysis revealed traits related to adhesion, bacteriocin production, and potential pathogenicity.
Abstract
Lacticaseibacillus rhamnosus is widely recognized for its health-promoting properties, which have led to its broad application in the production of food and dietary supplements. Nevertheless, although rare and typically limited to patients with underlying conditions, adverse effects have also been reported. In this study, we sequenced and characterized the genomes of seven L. rhamnosus strains isolated from blood. Using a hybrid approach that combined Illumina and Oxford Nanopore technologies, we obtained complete genomes ranging from 2.96 to 3.13 Mb, with a GC content of 46.7–46.8%. Comparative analyses with publicly available L. rhamnosus genomes revealed that these isolates were genetically related to strains from highly diverse origins, including plants, dairy products, dietary supplements, the gastrointestinal and genitourinary tracts, as well as blood and other clinical samples…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
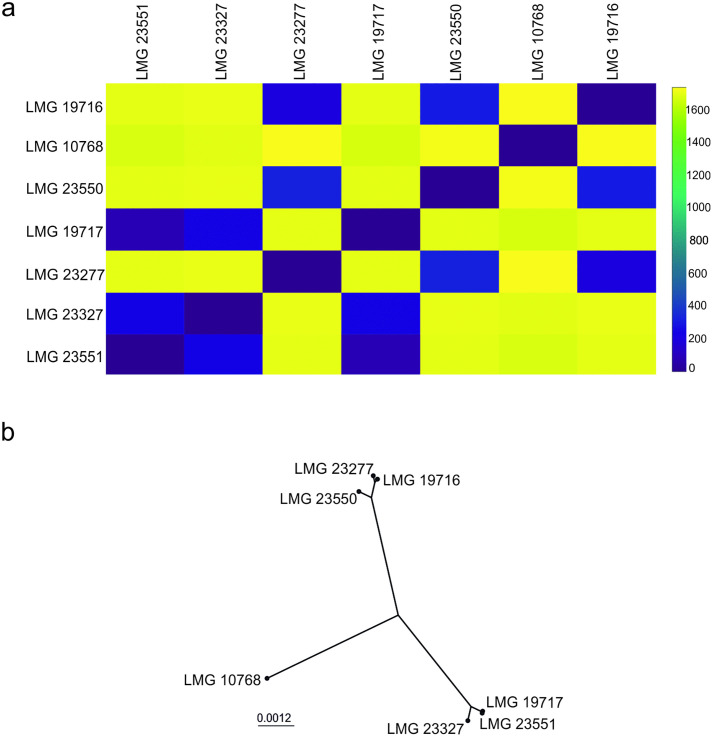 Fig 1
Fig 1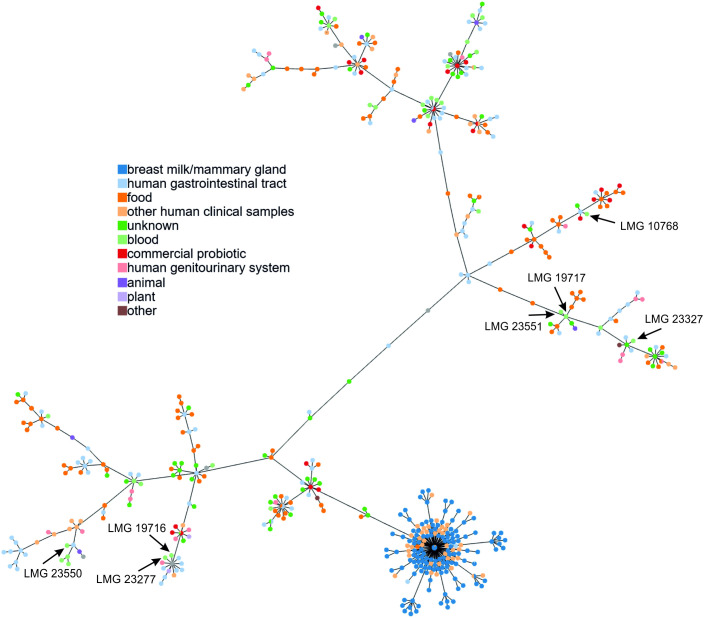 Fig 2
Fig 2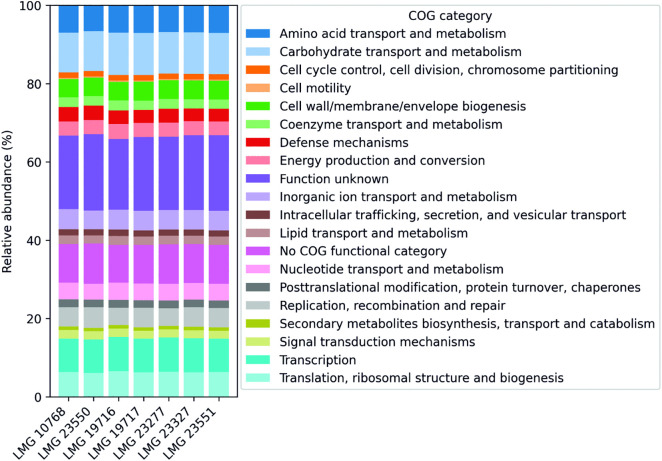 Fig 3
Fig 3- —http://dx.doi.org/10.13039/501100004281Narodowe Centrum Nauki
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProbiotics and Fermented Foods · Gut microbiota and health · Microbial Metabolites in Food Biotechnology
Introduction
Lactobacillus rhamnosus, recently reclassified as Lacticaseibacillus rhamnosus, is a widely studied bacterial species with important applications in the pharmaceutical and food industries [1,2]. Several strains, including GG, HN001, R0011, Oxy, Pen, E/N, and GR1, have demonstrated probiotic effects and are included in medical preparations with drug status [1,3–6]. Members of this species are gram-positive, homofermentative, non-spore-forming, and non-motile, and produce lactic acid from glucose [2,7,8]. As nomadic bacteria, L. rhamnosus inhabits diverse ecological niches, ranging from the human and animal gastrointestinal, genitourinary, and skin microbiota to fermented foods and environmental sources [2,8–10].
The beneficial effects of L. rhamnosus are mediated through multiple mechanisms, including intestinal colonization, inhibition of pathogenic microorganisms, and immune system modulation. Key contributing factors include the production of organic acids, bacteriocins, adhesion proteins, exopolysaccharides, and surface structures such as SpaCBA pili and lipoteichoic acids [11–19]. Clinically, L. rhamnosus strains have been associated with alleviating antibiotic-associated diarrhea, treating vaginal infections, improving oral and pulmonary health, and supporting immune function [5,20–22].
However, not all studies have reported consistent benefits. Some trials have found limited effects on antimicrobial-resistant colonization, microbiota restoration, and urinary tract infection prevention [23–26]. In addition, L. rhamnosus can pose risks in immunocompromised individuals, with documented cases of bacteremia, endocarditis, and other severe infections. These observations highlight that probiotic traits and pathogenicity are strain-specific, underscoring the need for precise strain-level identification in both research and clinical contexts [27–35].
While traditional methods for differentiating L. casei group bacteria were often labor-intensive and lacked reproducibility, advances in next-generation sequencing now allow comprehensive genome analyses for accurate identification and functional characterization [36–38]. In this study, we sequenced the complete genomes of seven blood-derived L. rhamnosus strains using hybrid sequencing method. The genomes were analyzed by core genome multilocus sequence typing (cgMLST) and subjected to functional genomic analyses to elucidate the unique features of these strains.
Materials and methods
Bacterial culture conditions and DNA isolation
L. rhamnosus strains (Table 1) were obtained from the Belgian Coordinated Collections of Microorganisms. Bacteria were cultured anaerobically in MRS broth (Difco) at 37°C. Genomic DNA was isolated and purified using the Genomic Mini AX Bacteria+ kit (A&A Biotechnology). The quality and concentration of genomic DNA were initially assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific) by measuring absorbance ratios at 260/280 nm and 260/230 nm. For more accurate quantification, DNA concentration was also measured using the Qubit 4.0 fluorometer (Thermo Fisher Scientific). DNA integrity was evaluated by electrophoresis on a 1% agarose gel stained with SimplySafe (Eurx) and visualized under UV light (Gel Doc XR + , Biorad).
Table 1: General features of complete genomic sequences obtained for the L. rhamnosus strains used in this study.
Library preparation and genomic sequencing
The concentration of genomic DNA was measured prior to library preparation using PicoGreen reagent (Life Technologies) and a Tecan Infinite instrument.
Library preparation was performed using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, USA) following the manufacturer’s protocol. Briefly, 125 ng of genomic DNA was mechanically fragmented using a Covaris E210 sonicator (Covaris), then end-repaired and dA-tailed. NEBNext® adapters were ligated, and the libraries were purified with AMPure XP beads (Beckman Coulter). Six cycles of PCR amplification were performed using NEBNext® Ultra™ II Q5® Master Mix and Illumina-compatible TruSeq CD Indexes, followed by a second bead-based purification. Libraries were pooled by equal mass. The quality and size distribution of the final pooled library was assessed using an Agilent Bioanalyzer and quantified by qPCR. High-throughput sequencing was carried out using MiSeq Reagent Kit v3 (600 cycles) chemistry (Illumina) obtaining at least 50x coverage of the bacterial genome.
High-quality genomic DNA (500 ng per sample) was used as input for library preparation. Sequencing libraries were prepared using the Rapid Barcoding Kit (Oxford Nanopore Technologies, UK) according to the manufacturer’s protocol. Each bacterial isolate was uniquely barcoded, and the resulting libraries were pooled equimolarly. The final pooled library was loaded onto a MinION device equipped with a MinION Flow Cell R9.4.1. (Oxford Nanopore Technologies) for whole genome sequencing.
Bioinformatics analysis
MiSeq reads were filtered using Cutadapt version 3.0 software [39]. Quality trimming was applied with a minimum Phred score of 25, and reads shorter than 15 bp after trimming were discarded. Quality control of sequencing data was performed with FastQC software [40]. Base calling for Oxford Nanopore sequences was carried out using Guppy version 6.1.2 (Oxford Nanopore) in high-accuracy mode with default Q-score settings. De novo assembly was performed using Unicycler version 0.4.7 [41] with default parameters. Genome assembly quality was assessed using QUAST [42] and CheckM [43].
The average nucleotide identity (ANI) of the obtained sequences was measured using the JSpecies Web Server [44] against selected reference genomes of L. rhamnosus, L. casei, L. paracasei, L. chiayiensis, and L. zeae. For cgMLST analysis, 615 publicly available L. rhamnosus genomic sequences were obtained from the NCBI Genome database using the NCBI Datasets tool. Genetic loci and alleles were identified, and a core gene set was determined. A schema was created using chewBBACA v3.3.2 [45] with default parameters: BLAST Score Ratio of 0.6, minimum length of 0, size threshold of 0.2, and translation table 11. A Prodigal training file was used for gene prediction. Loci present in at least 95% of the genomes were used for cgMLST analysis. A minimum spanning tree was generated using PHYLOViZ [46] and subsequently visualized in iTOL [47].
Prophage regions were identified with PHASTER using standard parameters, and classified into three groups (intact, questionable, and incomplete) based on the score values [48]. The presence of CRISPR/Cas modules was assessed using CRISPRCasFinder with the default settings [49]. Only sequences containing a complete set of cas genes were retained for further analysis. The COG evaluation was performed using the eggNOG-mapper software [50]. Potential probiotic characteristics and virulence factors were assessed using BLAST [51], ResFinder 4.6.0 [52], CARD [53], AMRFinderPlus [54], PathogenFinder [55], BAGEL4 [56], and ABRicate against Virulence Factor Database [57].
Availability of data
The L. rhamnosus genome sequences have been deposited in the GenBank database under accession numbers: CP136113.1 - CP136120.1. All data generated or analyzed in this study are included in the article and its supplementary information files.
Results and discussion
General characteristics of the L. rhamnosus genome
In the present study, involving seven strains of L. rhamnosus isolated from blood (Table 1), genomes were obtained using sequencing with two technologies — Illumina (MiSeq) and Oxford Nanopore (MinION). Previous studies have concluded that this hybrid approach appears to be the optimal choice for obtaining complete bacterial genomes [58,59].
Using Illumina short-read sequencing, numerous reads ranging from 1,201,885 (for LMG 23550) to 2,080,408 (LMG 19716), with an average length of 227.0 to 245.2 bp, were obtained for each strain. The base call accuracy, expressed by the average Q-score and Q30%, was above 35 and 88%, respectively. The results obtained were characterized by acceptable quality [60]. The average GC content for all reads in individual samples ranged from 46.63 to 46.68%. De novo assembly of Illumina reads resulted in 47–76 contigs per genome. The assembly quality, expressed as the N50 value, varied between 136,078 and 338,799 bp, with L50 values of 4–6 contigs. The final coverage obtained for individual samples ranged from 89.1 to 174.8. Detailed results of Illumina sequencing (MiSeq) are shown in Table 2.
Table 2: Summary of sequencing results from the Miseq platform (Illumina), obtained using QUAST software.
The application of Oxford Nanopore technology yielded reads, from 8,048 (LMG 23327) up to 190,157 (LMG 10768). The average read length for the samples tested ranged from 5,158.9 to 6,913.7 bp, with read N50 values between 10488 and 14472, indicating a substantial proportion of long reads suitable for resolving repetitive regions [61]. In the case of ONT, base call accuracy is usually much lower [62]; an average Q-score ranging from 11.9 to 12.5 was obtained for the tested samples. The approximate coverage for the tested strains varied from 18.6 (LMG 23327) to 441.5 (LMG 19717). The obtained results are included in Table 3.
Table 3: Summary of sequencing results from the MinION platform.
Finally, the reads obtained using both technologies were compiled using Unicycler software [46]. The final coverage for each genome ranged from 133.9 (LMG 23327) to 583.9 (LMG 19717) (Table 1). The obtained sequences met the basic minimum standards described by Riesco and Trujillo (2024) [63]. For all strains studied, full genomic sequences were obtained, with lengths ranging from 2,958,057 to 3,127,641 bp and GC content in the range of 46.7 to 46.8%. These values are consistent with previous literature reports describing genomes of strains belonging to L. rhamnosus [2,8].
In the analyzed genomes, 2,684 (LMG 19716) to 2,949 (LMG 23550) genes were identified, including sequences encoding rRNAs, tRNAs, ncRNAs, protein-coding genes, and pseudogenes (Table 1). Only for L. rhamnosus strain, LMG 19717 was a plasmid sequence of 46,439 bp obtained, in which 55 genes were identified. Similar plasmids were also detected in other L. rhamnosus strains, for example, hmr 1301 (98% sequence coverage, 99.98% sequence similarity), DM065 (84%, 99.54%), DM163 (84%, 99.56%), or PMC203 (81%, 99.54%). However, previous studies have shown that having plasmids is not a characteristic of all strains of this species [64].
Phylogenetic analysis
In the next part of the work, a phylogenomic analysis of the obtained L. rhamnosus genomes was performed. Comparison of the studied sequences to selected L. rhamnosus genomes using the ANIb algorithm showed very high similarity, exceeding 96%, confirming the species affiliation of the analyzed strains. It is expected that for strains belonging to the same species, the ANI should be at least 95 to 96% [65]. For genomes from other species of the L. casei group, ANIb similarity was below 80%.
cgMLST was used to determine the phylogenetic status of the isolates at the strain level [66]. The first step was to identify all genes and determine the set of core genes necessary to create a cgMLST schema. Schema construction was performed using all available L. rhamnosus genomes (615) with complete genome, chromosome, scaffold, and contig status. In the analyzed genomes, 9,390 loci were identified, for which 78,757 alleles were detected. The most variable loci included 85 alleles for tyrosine-protein kinase CpsD, 81 for S8 family serine peptidase, and 78 for extracellular matrix-binding protein EbhA. In addition, 5,101 loci were present in only one variant. The general characteristics of the genes used and their alleles are shown in S1 Fig.
A cgMLST scheme was then developed, containing loci present in at least 95%, 99%, and 100% of the analyzed L. rhamnosus genomes (S2 Fig). Due to the low quality of some sequences, it is recommended that a set of core genes found in at least 95% of genomic sequences be used for further analyses [67]. For the 615 L. rhamnosus genomes, 1,813 loci were used to develop the cgMLST95 scheme. The list of core genes and variants present in the analyzed L. rhamnosus genomes is included in S1 Table. Based on the resulting scheme, the seven genomes of the studied strains were analyzed both individually and together alongside the 615 genomic sequences deposited for the L. rhamnosus species.
The analysis showed that among the coding sequences in the studied genomes, 1,808–1,813 gene loci were identified, based on which the cgMLST scheme was developed. This represented about 61 to 67% of all genes detected in the studied strains. Analysis of the seven tested strains indicated that the genomes obtained for isolates LMG 23551, LMG 23327, and LMG 19717; and LMG 23550, LMG 23277, and LMG 19716 were relatively similar (Fig 1).
Analysis of the distances between seven L. rhamnosus strains based on core genome multilocus sequence typing (cgMLST) profiles obtained for 615 genomes.(A) Heatmap representing the allelic distance matrix for all samples in the dataset. The distances were computed by determining the number of allelic differences from the set of 1,778 core loci (loci that are not present in all samples are excluded from the calculation). (B) Core-genome neighbor-joining tree computed based on the multiple sequence alignment for the set of loci that constitute the core genome.
Based on the allelic profile achieved for the 622 L. rhamnosus genomic sequences, an MST was constructed using PHYLOViZ software (Fig 2). The L. rhamnosus genomes most similar to the sequences of the tested strains were then identified, with both the source of isolation and geographic location taken into account (S2 Table and S3 Fig).
Minimum spanning tree of L. rhamnosus strains based on cgMLST allelic profiles, constructed using PHYLOViZ.The arrows indicate isolates derived from blood: LMG 10768, LMG 19716, LMG 19717, LMG 23277, LMG 23327, LMG 23550, and LMG 23551. The node colors represent the source of bacterial isolation.
For strain LMG 23277, the most similar strains were bacteria isolated from the tongue (DM065 and DM163), stool (1001311H_170123_H11), and vaginal secretions (PMC203). Similar results were also obtained for strain LMG 19716, although the similarity to the above-mentioned strains was noticeably lower. Analysis of the genome of strain LMG 10768 revealed that the most similar isolates were ATCC 21052, LOCK900, and a strain derived from Egyptian cheese (CIRM-BIA 910). Slightly lower similarity was also observed with strains from a wide variety of sources, including commercial dietary supplements and pharmaceutical preparations. A very high degree of similarity was obtained for the two genomes presented in this paper (strains LMG 23551 and LMG 19717). These sequences were also similar to the genomes of isolates from dairy products from Georgia and Mongolia. The L. rhamnosus genomes most similar to the sequence obtained for strain LMG 23327 were those from bacteria derived from humans, including two strains isolated from urine. The last strain tested (LMG 23550) showed the highest homology with the INIA P344 isolate from infant feces (Spain), as well as several L. rhamnosus strains isolated from clinical or host-associated samples of intensive care patients (USA). Notably, this strain also showed similarity with three strains derived from blood [7].
In summary, the analysis of all L. rhamnosus genomes indicated that the studied strains showed high similarity to bacteria isolated from diverse sources, including the digestive tract, vagina, mouth, dairy products, and commercial probiotic supplements. It should also be emphasized that, due to the still insufficient number of genomic sequences from specific countries or regions, it was difficult to establish a definitive link between the studied strains and a particular geographical location.
As with the strains described by Nissilä et al. (2017), the genomic sequences obtained showed clear differences from that of the probiotic strain L. rhamnosus GG [7]. Due to its widespread use in health-promoting preparations, this strain has a global range. In some cases, correlation was observed with isolates from blood and other clinical samples, such as Lrh20, Lrh23, or Lrh30. These observations suggest the need for further research into the potential predisposition of these strains to colonize the human bloodstream.
Prophage-like elements as a source of strain-specific sequences in the genomes of L. rhamnosus
Previous studies have shown the widespread occurrence and extreme diversity of prophage-like sequences in the genomes of bacteria belonging to the L. casei group [68]. The presence of such sequences may play an essential role in the bacterial host and may also be important in shaping the entire ecological niche. Notably, in gut bacteria, prophages may regulate the diversity of the gastrointestinal microbiota, indirectly influencing the overall physiological state of the intestine [69,70]. It has also been shown that spontaneous prophage induction (SPI) can have important roles in biofilm formation and horizontal gene transfer and may shape bacterial pathogenic traits [71]. Despite their mobile nature, the high diversity of these sequences can also be relevant for genotyping bacteria at the strain level [38].
In the obtained genomes, prophage sequence identification was carried out using PHASTER software. A total of 22 sequences were found, including three intact sequences (score above 90), 11 questionable sequences (score between 61 and 90), and eight incomplete sequences (score below 60). The length of the detected sequences ranged from 4.5 to 56.2 kb, with GC content ranging from 43.08 to 49.01%. Depending on length, the sequences contained between 7 and 67 genes (Table 4).
Table 4: General characterization of prophage sequences identified in the genomes of individual bacterial strains obtained in this study.
For strain LMG 10768, two prophage sequences were identified, one of which (R3_1) was almost identical to sequences from blood isolates Lrh20 and Lrh23, suggesting possible associations between certain prophages and strains of clinical origin. In contrast, the second sequence (R3_2) appeared to be strain-specific, with only fragmentary homologs in other genomes. The chromosome of LMG 23550, was particularly rich in prophage sequences, which represented nearly 6% of the total genome. One sequence, only 4.5 kb long and encoding eight proteins (including an IS3 family transposase), was found in many strains of both L. rhamnosus and in closely related species. The remaining prophages displayed only partial similarity to those found in other solates, indicating that analyzed strain harbors unique mobile elements.
Only one prophage-like sequence was identified in L. rhamnosus LMG 19716. Similar prophages with 91–94% similarity were present in the genomes of L. rhamnosus (k32, 51B, LB, RSI3) and L. casei (FBL6) strains. This suggests that certain phage elements may circulate within closely related species. In the L. rhamnosus LMG 19717 genome, three prophages were detected. Two (R18_2 and R18_3) were widely distributed among L. rhamnosus strains, whereas the third sequence (R18_1) contained fragments that were unique to this strain. The most similar sequence, with 99.92% similarity and 94% sequence coverage, was found in the L. rhamnosus LMG 23551 genome obtained in this study. In another strain (LMG 23277), three prophages were identified that were widespread across the species but often displayed incomplete coverage, highlighting structural variation even in conserved sequences. Finally, the last two strains (LMG 23327 and LMG 23551) carried multiple prophages, some of which (e.g., R24_1 and R26_2) were broadly distributed across L. rhamnosus and L. paracasei strains from diverse environments, whereas others were more restricted (Table 4).
Overall, the analysis of prophage-like sequences confirmed the broad occurrence of such elements in L. rhamnosus genomes, comprising both universal and strain-specific sequences. This variability not only highlights the dynamic nature of the L. rhamnosus mobilome but also constitutes a source of specific sequences enabling the identification of individual isolates at the strain level [38,68,72].
Characterization of CRISPR-Cas systems in analyzed strains
CRISPR-Cas systems are also essential components of bacterial genomes. Given the high variability of motifs referred to as spacers, these sequences can be used for genotyping bacteria at the strain level. Bacterial identification methods based on CRISPR loci are especially relevant for the specific detection of pathogens in clinical samples and food [73–75]. However, a limitation of this approach is that the presence of CRISPR motifs is not universal. It is estimated that CRISPR sequences are observed in only about 40% of bacterial genomes [76].
In the present study, complete CRISPR modules categorized as the CAS-Type IIA subtype were detected in four of the seven genomes examined. For strains LMG 23327, LMG 23551, LMG 19717, and LMG 10768, 28, 37, 27, and 40 spacers were identified, respectively, with identical repeat sequences of 36 nucleotides in length located between them. Genes encoding CRISPR-associated (Cas) proteins — Cas1, Cas2, Cas9, and Csn2 — were also detected. A detailed analysis of CRISPR modules in L. rhamnosus genomes revealed the presence of similar sequences in other strains. For three of the four strains tested with the CRISPR module, some spacer sequences were strain-specific (S3 Table).
An alignment of the CRISPR module obtained for strain LMG 10768 with L. rhamnosus genomes showed the highest similarity to clinical isolate 186_LRHA. In the genome of this strain, 59 spacer sequences were detected; however, compared to LMG 10768, spacer sequences 5, 39, and 40 were missing. The DSM 14870 and TK-F8B strains also shared high similarity, each with 27 identical spacers. Notably, the last spacer was identified in plasmid sequences of L. plantarum and L. kefiri.
For LMG 23327, six strains had identical CRISPR modules (QAULRN2, RAB2019A, Fmb14, 1001095st1_F3_1001254B_151014, 1001095st1_F6_1001095A_150126, and UW_DM_LACCAS2_1), all sharing 28 spacers. In addition, CRISPR sequences with very high similarity were also detected in many other L. rhamnosus genomes. Individual spacer analysis revealed that spacer 5 was present in phage Lrm1, spacer 26 in L. plantarum plasmids, while spacer 27 was identified in the genomes of phages T25, R9.3, C3.1, and C4.1. Spacer 28 was commonly found in many phages of the L. casei group, including BH1, C3.1, C4.1, R3.1, R9.2, R29.1, R18.1, and R26.14, as well as in several sequences of Caudoviricetes sp. and Siphoviridae sp. obtained from metagenomic studies [77].
The CRISPR array obtained from the genome of LMG 19717 contained 27 spacer sequences, 26 of which were also found in the genomes of strains LMG 23551, UMB13151A, and LR5. Some spacer sequences were further identified in L. rhamnosus strains SD4, SD11, Fmb14, UMB0004, WHH1155, LMG 23327, and others. In addition, some sequences, specifically spacers 4, 26, and 27, were detected in mobile elements, including L. plantarum plasmids and L. casei group phages [68,78].
For the final strain (LMG 23551), BLAST analysis showed highest similarity to UMB131A and LMG 19717, with 26 identical spacers out of the 37. Similar sequences were also detected in LR5, Fmb14, UMB0004, SD4, SD11, LMG 23327, and WHH1155, originating from various sources (dairy-fermented products, feces, saliva, and blood). Several spacers have been identified in the plasmids of species formerly classified within the genus Lactobacillus, as well as in Pediococcus and Lactococcus. Notably, spacers 4 and 29 were observed in the phage sequences of Lrm1 and BH1 [78,79].
In conclusion, CRISPR modules, particularly spacer sequences, appear to be essential components of bacterial genomes, aiding in the reconstruction of the genetic history of individual bacterial strains. The identification of similar sequences across different isolates suggests a close evolutionary relationship and a shared lineage. Furthermore, the enormous diversity of these motifs may facilitate the development of strain-specific molecular probes for bacterial identification. Among the strains studied, in some cases, genomes were identified with an identical set of spacers, indicating a possible close genetic relationship. In addition, conserved sequences were observed across the tested strains. Interestingly, the complete set of spacer sequences identified in this study was absent from the CRISPR module of the L. rhamnosus GG genome. This finding suggests that, despite its widespread distribution, L. rhamnosus GG is not closely related to the blood-derived isolates examined. However, spacer sequences originating from the L. rhamnosus GG genome were present in the prophage sequences identified in the genomes of strains LMG 23277 and LMG 23327.
Functional analysis of genomic sequences
The genes identified in the L. rhamnosus genomes were classified into 19 functional categories of clusters of orthologous groups (COGs) (Fig 3 and S4 Table). For all strains tested, approximately 10% of the genes were not assigned to any category. Among the genes with a specific COG class, the largest group (about 10.2–10.8%) consisted of sequences encoding proteins related to the transport and metabolism of carbohydrates, amino acids (6.6–7%), transcription (8.5–8.8%), and translation (6–6.5%). Nearly 20% of the genes were sequences classified in the category for genes of undetermined function. This emphasizes the need for further research to better understand the identified genes. Notably, the number of genes in each COG category was fairly similar across the strains studied. The low variation between isolates may reflect similar adaptive strategies for survival in a specific ecological niche [8].
Relative abundance of COG (Clusters of Orthologous Groups) categories across the analyzed genomes.Each COG category is represented by a distinct color, enabling comparison of functional category distributions among genomes.
Analysis performed with ResFinder 4.6.0, CARD, and AMRFinderPlus software did not indicate the presence of genes associated with antibiotic resistance. Only in the case of L. rhamnosus LMG 10768 was a sequence encoding the MFS transporter detected — a protein responsible for transporting ions, sugar phosphates, drugs, neurotransmitters, nucleosides, amino acids, and peptides across cytoplasmic or internal membranes [80,81]. This finding is important for potential antibiotic therapy in bacteremia caused by L. rhamnosus strains. Moreover, PathogenFinder, available through the Center for Genomic Epidemiology resources, indicated that regardless of the isolation source, the strains studied cannot be classified as human pathogens [55]. Similarly, screening against the Virulence Factor Database (VFDB) using ABRicate did not reveal the presence of known virulence determinants [57]. In line with previous studies involving clinical L. rhamnosus strains, it remains difficult to definitively identify the genes that determine the ability of these isolates to cause bacteremia. Based on scientific reports, it can be speculated that factors such as adhesion-related proteins, modified EPS clusters, pilus genes, or the phenomenon of SPI may play a role, among others [7,35,71,82]. However, these elements provide physiologically relevant functions that facilitate colonization of the human gastrointestinal tract and are present in many strains that are considered as safe.
Comparative analysis revealed the presence of genes encoding sortases and the proteins they recognize, which contain the LPXTG motif (S5 Table). These proteins are considered important adhesion factors that affect the ability of bacteria to bind to intestinal epithelial cells [83]. Studies have also shown that these proteins are very important pathogenicity factors [84,85]. Thus, what may appear to be a beneficial, health-promoting feature could instead contribute to infection, particularly in patients with health issues, for whom probiotic use should be limited [27,86]. Similar to the strains described by Nadkarni et al. (2014) and Nissilä et al. (2017), spaFED clusters encoding pilus proteins were detected in the analyzed genomes [7,82]. Their role in bacterial adhesion has been well documented [87,88]. The second pilus module (spaCBA) was present only in L. rhamnosus strain LMG 19717 and was interestingly located on a plasmid (CP136117.1). Analysis showed that spaCBA is less common among L. rhamnosus strains, although a similar sequence was also observed in the genomes and plasmids of L. paracasei [89]. Due to the presence of mobile elements near the spaCBA genes, it is possible that this module was acquired via horizontal gene transfer.
The production of EPS is another crucial factor that influences adhesion properties and biofilm formation. In the genomes of the strains studied, many sequences related to EPS biosynthesis were detected, with their roles in the production of these compounds already established [14]. In all genomes examined, at least one complete cluster encoding genes related to EPS production was present (S5 Table). It should be noted that these modules exhibit high variability. In some cases, genes encoding proteins with specific functions in EPS synthesis showed no sequence similarity to genes previously described for L. rhamnosus GG. This may be due to the presence of mobile elements within these sequences, such as transposase enzymes [7].
Genetic determinants responsible for the synthesis of bacteriocins were the final element investigated in the L. rhamnosus genomes. These proteins exhibit bacteriostatic properties, making them relevant for the direct competition between L. rhamnosus strains and other bacteria within the same ecological niche [90,91]. Previous studies have shown that bacteria of this species can synthesize bacteriocins such as carnocine CP52, enterocin X chain beta, and class II bacteriocins with a double-glycine leader peptide [92]. Similar genetic clusters were also detected in all L. rhamnosus genomes analyzed in this study (S5 Table).
Conclusions
In this study, we obtained the complete genome sequences of seven L. rhamnosus blood isolates using a hybrid short-read/long-read sequencing approach. Hybrid sequencing is an optimal strategy for obtaining complete bacterial genomes, enabling precise characterization and identification of specific strains relevant to industry or medicine. Comparative analysis using cgMLST demonstrated phylogenetic relationships with previously characterized L. rhamnosus strains, confirming the utility of this method for strain-level differentiation of L. rhamnosus. Based on the results, it can be concluded that the strains studied do not show a clear similarity with only clinical isolates. In addition, they are not closely related to commercial strains (such as L. rhamnosus GG), which are used in dietary supplements and as food additives. The analysis revealed a wide diversity of strains in terms of both the source of isolation and geographic origin, which exhibited similarity to the L. rhamnosus strains studied. Similar conclusions can be drawn from analyses based on prophage sequences and CRISPR arrays. Given the very high genetic diversity, these modules appear to be a valuable complement to genomic analyses, such as cgMLST. Genomic analysis also identified numerous genes associated with traits commonly linked to probiotics. Some of these genes are also present in pathogenic bacteria and have been implicated in microbial virulence. These findings emphasize that the mere presence of such genes in a genome does not necessarily indicate beneficial effects and should be interpreted carefully when characterizing bacterial strains.
Supporting information
S1 FigcgMLST schema data summary, including.(A) Distribution of the number of alleles for all loci, (B) Distribution of the allele mode size (most frequent allele size) for all loci, and (C) Total number of alleles and the minimum, maximum, and median allele size per locus. Loci and alleles were identified in 615 L. rhamnosus genomes (complete genomes, chromosomes, scaffolds, and contigs).(TIF)
S2 FigAnalysis of the loci number in cgMLST, assuming that a given gene is present in 95, 99, and 100% of the analyzed genomes.The observation was performed using 615 L. rhamnosus genomic sequences (including complete genomes, chromosomes, scaffolds, and contigs).(TIF)
S3 FigMinimum spanning tree of L. rhamnosus strains based on cgMLST allele profiles.Distances were calculated with cgmlst/dists, and the MST was generated using NetworkX (Kruskal’s algorithm). The resulting tree was exported in Newick format and visualized with iTOL. Concentric circles around strains indicate metadata: the inner circle shows geographic origin, and the outer circle shows the isolation source, both encoded with colors as specified in the legend. Strains analyzed in this study are highlighted in green.(PDF)
S1 TableOccurrence of individual genes and alleles in the 615 L. rhamnosus genomes.Loci present in at least 95% of the analyzed genomic sequences were used to construct the cgMLST scheme.(XLSX)
S2 TableDistance matrices for all tested strains, constructed from a node selection of the cgMLST L. rhamnosus dataset.(XLSX)
S3 TableBasic information on CRISPR/Cas sequences identified in the genomes of L. rhamnosus strains LMG 10768, LMG 19717, LMG 23327 and LMG 23551.(XLSX)
S4 TableFunctional study of L. rhamnosus genome sequences, presenting the distribution of genes within individual COG categories.(XLSX)
S5 TableAnalysis of the occurrence of genes encoding proteins related to adhesion, production of exopolysaccharides, and bacteriocins in the genomes of the L. rhamnosus strains studied.(XLSX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Capurso L. Thirty Years of Lactobacillus rhamnosus GG: A Review. J Clin Gastroenterol. 2019;53(1):S 1–41. doi: 10.1097/MCG.0000000000001170 30741841 · doi ↗ · pubmed ↗
- 2Zheng J, Wittouck S, Salvetti E, Franz CMAP, Harris HMB, Mattarelli P, et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int J Syst Evol Microbiol. 2020;70(4):2782–858. doi: 10.1099/ijsem.0.004107 32293557 · doi ↗ · pubmed ↗
- 3Miao X, Jiang Y, Kong D, Wu Z, Liu H, Ye X, et al. Lactobacillus rhamnosus HN 001 Ameliorates BEZ 235-Induced Intestinal Dysbiosis and Prolongs Cardiac Transplant Survival. Microbiol Spectr. 2022;10(4):e 0079422. doi: 10.1128/spectrum.00794-22 35862958 PMC 9430965 · doi ↗ · pubmed ↗
- 4Foster LM, Tompkins TA, Dahl WJ. A comprehensive post-market review of studies on a probiotic product containing Lactobacillus helveticus R 0052 and Lactobacillus rhamnosus R 0011. Benef Microbes. 2011;2(4):319–34. doi: 10.3920/BM 2011.0032 22146691 · doi ↗ · pubmed ↗
- 5Ruszczyński M, Radzikowski A, Szajewska H. Clinical trial: effectiveness of Lactobacillus rhamnosus (strains E/N, Oxy and Pen) in the prevention of antibiotic-associated diarrhoea in children. Aliment Pharmacol Ther. 2008;28(1):154–61. doi: 10.1111/j.1365-2036.2008.03714.x 18410562 · doi ↗ · pubmed ↗
- 6Nader-Macías MEF, De Gregorio PR, Silva JA. Probiotic lactobacilli in formulas and hygiene products for the health of the urogenital tract. Pharmacol Res Perspect. 2021;9(5):e 00787. doi: 10.1002/prp 2.787 34609059 PMC 8491456 · doi ↗ · pubmed ↗
- 7NissiläE, Douillard FP, Ritari J, Paulin L, Järvinen HM, Rasinkangas P, et al. Genotypic and phenotypic diversity of Lactobacillus rhamnosus clinical isolates, their comparison with strain GG and their recognition by complement system. P Lo S One. 2017;12(5):e 0176739. doi: 10.1371/journal.pone.0176739 28493885 PMC 5426626 · doi ↗ · pubmed ↗
- 8Douillard FP, Ribbera A, Kant R, PietiläTE, Järvinen HM, Messing M, et al. Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. P Lo S Genet. 2013;9(8):e 1003683. doi: 10.1371/journal.pgen.1003683 23966868 PMC 3744422 · doi ↗ · pubmed ↗