Multiplex PCR assay for the rapid detection of Klebsiella pneumoniae pathotypes
Sanika Mahesh Kulkarni, Jobin John Jacob, T. Praveen, V. Aravind, R. Subbulakshmi, S. Preethi, Binesh Lal, Karthik Gunasekaran, Abi Manesh, Shraddha M. Karve, J. Sudarsana, Sanjay Bhattacharya, Anand Shah, Savitha Nagaraj, Priyadarshini Padaki, S. Jayakumar, Renu Mathew

TL;DR
This paper introduces a new PCR test that can quickly and accurately detect different types of Klebsiella pneumoniae, including drug-resistant and highly virulent strains.
Contribution
The study presents a novel multiplex PCR assay that simultaneously detects multiple pathotype markers in Klebsiella pneumoniae with high specificity and sensitivity.
Findings
The m-PCR assay achieved 100% specificity when compared to whole-genome sequencing data.
The assay successfully detected all target genes without cross-amplification in control strains.
It demonstrated high sensitivity with efficient amplification from minimal DNA input.
Abstract
Introduction. Klebsiella pneumoniae (Kp) is a major cause of nosocomial infections, with its evolving pathotypes including multidrug-resistant, hypervirulent (hvKp) and convergent strains posing significant diagnostic and treatment challenges due to combined antimicrobial resistance and virulence. Gap Statement. While there is a pressing requirement for thorough detection of Kp pathotypes, current assays in resource-limited environments are unable to effectively focus on essential carbapenemase and hypervirulence genes with the necessary reliability and precision. Aim. To develop and validate a multiplex PCR (m-PCR) assay capable of simultaneously detecting Kp isolates including those carrying partial or full virulence markers, alongside antimicrobial resistance. Methodology. In this study, an m-PCR assay was designed and optimized for the simultaneous detection of key biomarkers…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
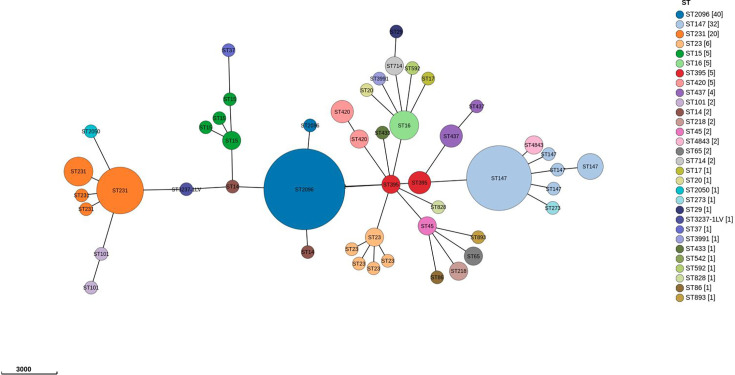 Fig. 1
Fig. 1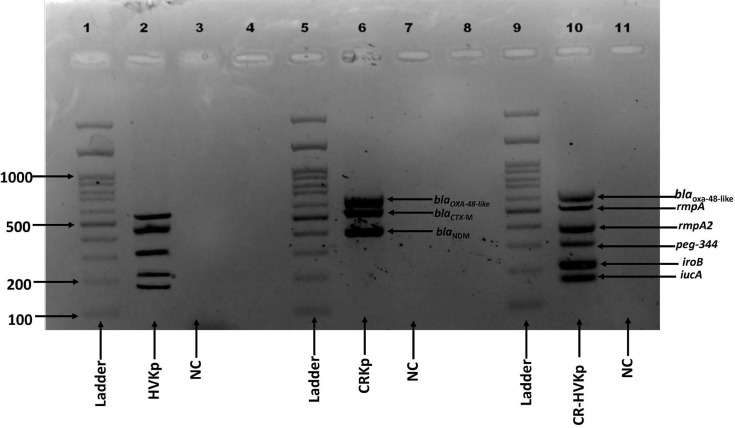 Fig. 2
Fig. 2| Target (phenotype) | Gene target | Sequence | Product size |
|
|---|---|---|---|---|
| Hypervirulence |
| CTAAAGCAGTTAACTGG | 530 bp | 1.2 |
| CATCTTTCATCAACCATTT | ||||
|
| AAGAGTATTGGTTGATAGCCGGA | 470 bp | 1.2 | |
| AGGTATTTGATGTGCACCATTTT | ||||
|
| TGGTTAACTCCACTTTTGCCGT | 170 bp | 1.2 | |
| ACCTTTTACGTTCCAGTACG | ||||
| Carbapenemase producing |
| GGCGTAGTTGTGCTCTGGAA | 630 bp | 1.2 |
| TCTTTTGTGATGGCTTGGCG | ||||
|
| TTTGATCGTCAGGGATGGCG | 401 bp | 1.2 | |
| TGATCAGGCAGCCACCAAAA | ||||
|
|
|
|
|
|
| Additional genes (hypervirulence) |
| TGGGGATAAGTGTGATAAGT | 313 bp | 1.2 |
| TTACCGTGTTTTATAGCTGG | ||||
|
| ATCTACCCTCCGCTCGGAGT | 219 bp | 1.2 | |
| GTCGTTTTCAAGAATGCTCA | ||||
| Carbapenemase producing [ |
| CGTCTAGTTCTGCTGTCTTG | 798 bp | 100 |
| CTTGTCATCCTTGTTAGGCG | ||||
| ESBL producer |
| SCNATGTGCAGYACCAGTAARG | 560 bp | 1.2 |
| CCGCRATATGRTTGGTGGTG |
| Temp. (°C) | Time | Cycles | |
|---|---|---|---|
|
| 95 | 10 min | 35 |
|
| 94 | 30 sec | |
|
| 50 | 1 | |
|
| 72 | 1 min | |
|
| 72 | 10 min | |
|
| 4 | ∞ |
- —http://dx.doi.org/10.13039/501100001411 Indian Council of Medical Research
- —http://dx.doi.org/10.13039/501100005918 Christian Medical College, Vellore
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Nosocomial Infections in ICU · Bacterial Identification and Susceptibility Testing
Introduction
Klebsiella pneumoniae (Kp), a Gram-negative, non-motile bacterium belonging to the order Enterobacterales, is known for its rapid dissemination, persistence and phenotypic diversity [12]. In hospital settings, Kp poses a significant threat, particularly to critically ill patients in intensive care units, immunocompromised individuals, the elderly and neonates [34]. It is a frequent cause of severe infections, including pneumonia, urinary tract infections, sepsis, wound infections and meningitis. Clinically, Kp is classified into two primary pathotypes: classical (cKp), which is responsible for nosocomial infections, and hypervirulent (hvKp), which is linked to community-acquired infections. cKp is further evolved into multidrug-resistant (MDR-Kp) or carbapenem-resistant (CR-Kp) strains based on their antimicrobial resistance (AMR) profiles [58].
The prevalence of MDR-Kp in clinical settings has risen dramatically in recent decades. Studies indicate that approximately one-third of hospital-acquired Gram-negative infections are caused by Kp [9]. In India, surveillance data from a multi-hospital network revealed that Kp accounts for 18% of bloodstream infections, with 57% of isolates exhibiting carbapenem resistance [10]. Treating these infections is particularly challenging due to their resistance to most antibiotics, including last-line therapies. Consequently, the World Health Organization has classified CRKp and extended-spectrum β-lactamase (ESBL)-producing strains as critical-priority pathogens [11]. The frequent acquisition of virulence factors, metal resistance and other horizontally transferred genetic elements further complicates the management of Kp infections [1213].
Beyond nosocomial settings, hvKp has emerged as a significant community-acquired pathogen over the past two decades. Historically, hvKp strains exhibited a hypermucoviscous (hmv) phenotype, which was qualitatively assessed using the ‘string test’ [14]. This phenotype, along with enhanced virulence, stems from the overproduction of capsular polysaccharides and siderophores, driven by genes on the virulence plasmid (pLVPK), such as rmpADC, rmpA2, iroBCDN and iucABCDiutA [15]. In recent years, hvKp has evolved into MDR-hvKp by acquiring MDR plasmids. Conversely, MDR-Kp strains have integrated virulence plasmids, giving rise to convergent clones that exhibit both resistance and hypervirulence [16]. These strains are most commonly reported in China, Southeast Asia and East Asia, with increasing cases documented in Europe, the USA and other Western countries [1517]. In India, a study from a tertiary care hospital identified 8% of isolates as belonging to this convergent pathotype [18]. The emergence of CR-hvKp, harbouring resistance genes such as blaKPC, blaNDM and blaOXA-48-like, has severely limited treatment options, often resulting in untreatable infections. The rising prevalence of MDR-hvKp in hospital settings is a pressing public health concern [1922]. Differentiating Kp pathotypes is thus essential for assessing infection severity and devising effective treatment strategies.
Traditionally, hvKp identification relied on a positive string test (string >5 mm) and susceptibility to commonly used antibiotics. However, most convergent clones lack hmv and test negative in the string test, limiting their reliability as a specific marker for hvKp [2324]. Genetic determinants of hypervirulence, such as aerobactin (iucA), salmochelin (iroB), metabolic transporter (peg-344) and regulator of mucoid phenotype (rmpA/rmpA2), have emerged as more robust biomarkers for PCR-based detection [2526]. Not all hvKp strains carry the same set of virulence genes, and frameshift mutations in rmpA/rmpA2 can reduce the sensitivity of PCR assays [2628]. For this study, hvKp was defined as isolates carrying all five canonical virulence genes: rmpA, rmpA2, iucA, iroB and peg-344, which are typically co-located on the pLVPK-like virulence plasmid to ensure accurate identification of truly convergent clones [2329]. Isolates carrying only a subset of these markers (e.g. iucA, rmpA and rmpA2) are referred to as ‘virulence marker–positive cKp’ rather than hvKp. Given the urgent need for a rapid, comprehensive molecular tool to identify and monitor convergent strains carrying both resistance and virulence determinants, we evaluated multiple genotypic biomarkers to distinguish all three Kp pathotypes: CRKp, hvKp and CR-hvKp. Using these biomarkers, this study developed a multiplex PCR (m-PCR) assay to accurately detect carbapenem-resistant hvKp and inform tailored treatment strategies for affected patients.
Methods
Genome sequence data retrieval
We have leveraged whole-genome sequencing (WGS) data available at NCBI pathogen detection (https://www.ncbi.nlm.nih.gov/pathogens/) to identify carbapenem-resistant hvKp genomes. This database integrates genome sequences and annotates AMR and virulence genes present in bacterial genomes. Among the 52,502 assembled genomes of Kp available in the database (dated 3 July 2023), we filtered and downloaded genomes with specific virulence markers (rmpA/rmpA2/iucA/iroB/peg-344) in NCBI pathogen detection and screened for 12,846 genomes (Table S3, available in Supplementary Material 1). In this study, hvKp was defined strictly as isolates carrying all five canonical markers (rmpA, rmpA2, iucA, iroB and peg-344). Subsequently, genomes carrying carbapenemase genes such as blaNDM and/or blaOXA-48 family, specific to Indian clinical contexts, were selected using various filter options within the database (https://www.ncbi.nlm.nih.gov/pathogens/refgene/). MLST, virulence genes, resistance and virulence scores of genome assemblies were examined using Kleborate v2.4.1 (https://github.com/klebgenomics/Kleborate/releases/tag/v2.4.1) and BLASTn against reference genes downloaded from the Institut Pasteur database (https://bigsdb.pasteur.fr/cgi-bin/bigsdb/bigsdb.pl?db=pubmlst_klebsiella_seqdef&page=downloadAlleles).
Primer design
For screening carbapenem-resistant hvKp, the coding regions of genes associated with hypervirulence (rmpA, rmpA2, iucA, peg3-44 and iroB) were extracted from the genome assemblies (n=8177) through BLASTn searches locally, employing a sequence identity threshold of >80% and a coverage threshold of >80%. All reported blaNDM and blaoxa-48-like variant sequences in the NCBI reference gene catalogue (https://www.ncbi.nlm.nih.gov/pathogens/refgene/) were downloaded. Subsequently, the resulting FASTA files were aligned to reference sequences using the MAFFT program (https://github.com/GSLBiotech/mafft). The m-PCR primers were designed for the primary assay, targeting conserved regions of rmpA, rmpA2, iucA, blaNDM and blaOXA-48-like. Additional primers were developed/included for peg-344, iroB, blaKPC and blaCTX-M allowing for flexibility in panel customization. Primer design was performed using the Thermo Fisher OligoPerfect Primer Designer (https://apps.thermofisher.com/apps/oligoperfect/). Selection criteria included a G+C content close to 50 mol%, a ΔTm of <2 °C and a length of 18–22 bases. Before empirical testing, these primers were initially evaluated in silico against all target and non-target sequences using NCBI primer-blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and in silico PCR (https://insilico.ehu.es/PCR/). Specific primers and assays that target blaKPC and blaCTX-M were adapted from previously published data [3031]. All oligonucleotide primer sequences used in this study are listed in Table 1 and were synthesized by Integrated DNA Technologies Pvt Ltd, Singapore. Reference standard strains, as well as clinical isolates, were used for primer optimization and in vitro testing.
Characterization of bacterial isolates
Kp strains (n=150) used in this study were clinical isolates collected between 2022 and 2023 at the Department of Clinical Microbiology, Christian Medical College, Vellore (n=47), and from multiple centres across India, including Baby Memorial Hospital, Kozhikode (n=76); Zydus Hospital, Ahmedabad (n=10); Saveetha Medical College, Chennai (n=5); St. John’s Medical College, Bengaluru (n=5); BCMCH, Thiruvalla (n=3); and TATA Medical Center, Kolkata (n=4) Supplementary Material 1 (Table S4). These isolates were sub-cultured on MacConkey agar at recommended culture conditions. Isolates were identified and confirmed as Kp by biochemical tests and MALDI TOF MS (VITEK^®^ MS, bioMérieux). Screening for the hmv phenotype was conducted via the string test, following the described semi-quantitative method [1]. A positive string test was defined by the formation of a mucoid string exceeding 5 mm in length upon contact with an inoculation loop.
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing was performed by the Kirby–Bauer disc diffusion method according to CLSI 2022 and 2023 guidelines [3233]. The antimicrobial agents tested included meropenem (10 µg) and ertapenem (10 µg). Controls utilized for the testing included Escherichia coli ATCC 25922, Enterococcus faecium ATCC 29212 and Pseudomonas aeruginosa ATCC 27853.
DNA extraction and WGS
The study isolates (n=150) were cultured in LB broth (Luria Bertani Broth) at 37 °C. Total genomic DNA was extracted from pelleted cells using the Wizard DNA purification kit (Promega, WI, USA). The concentration of extracted DNA was determined using NanoDrop One spectrophotometry (Thermo Fisher Scientific, MA, USA) and Qubit 3.0 fluorometry (Life Technologies, CA, USA), and the samples were stored at −20 °C until further analysis.
A sequencing library was prepared using the Nextra DNA Flex library preparation kit (Illumina, San Diego, CA). Initially, genomic DNA (1 µg) was fragmented, and adapters were ligated to the ends. The ligated DNA libraries underwent size selection, and the resulting products were PCR amplified with index primers following the manufacturer’s instructions. Subsequently, the paired-end library was sequenced on a NovaSeq 6000 platform (Illumina, USA) at Unipath Specialty Laboratory Limited, Ahmedabad, India, generating 2×150 bp reads. Sequencing reads with a PHRED quality score below 20 were discarded, and adapters were trimmed using cutadapt v1.8.1. Quality assessment was performed using MultiQC (https://github.com/ewels/MultiQC).
Comparative genome analysis
The resulting high-quality reads were then subjected to assembly using Unicycler (https://github.com/rrwick/Unicycler). MLST, virulence genes, resistance and virulence scores were examined using Kleborate (https://github.com/klebgenomics/Kleborate/releases/tag/v2.4.1). The assembled Klebsiella genomes were typed using the Ridom SeqSphere+ platform based on the core genome MLST (cgMLST) scheme for Klebsiella pneumoniae/variicola/quasipneumoniae available at https://www.cgmlst.org/ncs/schema/Kpneumoniae2267/. A total of 2,358 loci were selected, and alleles were called using the chewBBACA algorithm (https://github.com/B-UMMI/chewBBACA). Minimum spanning trees (MSTs) were constructed and visualized using GrapeTree (https://achtman-lab.github.io/GrapeTree/MSTree_holder.html) based on the cgMLST. Tree nodes were positioned through dynamic rendering, and node style was adjusted by fine-tuning the node size and kurtosis. Nodes were coloured by the pathotype of the isolates, and node sizes were drawn proportionally to the number of isolates.
Testing and optimization of m-PCR assay
m-PCR primer optimization
To optimize the m-PCR assay, each set of primers was individually tested in monoplex PCR reactions to amplify specific gene targets. Initially, combinations of two genes were tested, and the most efficient pair was selected for the initial double PCR. Subsequently, additional primer pairs were sequentially incorporated to form triple PCR reactions. This iterative process was repeated until the final m-PCR assay was successfully developed. The core targets of the assay included rmpA, rmpA2, iucA, blaOXA-48-like and blaNDM, while iroB, peg-344, blaKPC and blaCTX-M (included to enable detection of ESBL-producing strains) were standardized and can be incorporated optionally based on specific requirements. The blaCTX-M primer set was adapted from Lartigue et al. [31], retaining the reverse primer and modifying the forward primer as mentioned in Table 1.
m-PCR cyclic condition optimization
The PCR reaction was prepared using the Qiagen Multiplex PCR kit, comprising HotStarTaq DNA polymerase, Multiplex PCR Buffer and a Q-Solution. Each reaction was set up in a 20 µl volume, containing 10 µl of mastermix, 2 µl of Q-Solution and 6 µl of pooled primers (with 2 µl of each forward and reverse primer for 5 genes combined to make a total of 20 µl, to which 80 µl of nuclease-free water was added). Additionally, 2 µl of DNA template was added to each reaction. The PCR amplification was carried out using a Veriti thermal cycler (Applied Biosystems Inc., Foster City, CA). Annealing temperatures ranging from 46 °C to 56 °C were tested to optimize the amplification conditions. Subsequently, 5 µl of the reaction mixture was loaded onto a 2% agarose gel and electrophoresed at 130 V for 45 min to visualize the resulting amplicons.
PCR specificity and sensitivity
The specificity of the m-PCR assay was evaluated using 150 clinical isolates of Kp and 12 bacterial strains, including reference strains of Klebsiella quasipneumoniae ATCC 700603, Kp ATCC BAA-1705 and BAA-1706, Escherichia coli ATCC 35218, Pseudomonas aeruginosa ATCC 27853, Acinetobacter baumannii ATCC 19606, Staphylococcus aureus ATCC 43300 and Streptococcus pneumoniae ATCC 17815, as well as strains of Salmonella Typhi, Shigella sp., Morganella morganii, Serratia marcescens and Proteus mirabilis from IHMA. Genomic DNA extraction was carried out following the procedure outlined in the section ‘DNA extraction and WGS’, and PCR amplification was performed as described in the section ‘m-PCR cyclic condition optimization’.
A tenfold serial dilution procedure was adapted to assess the sensitivity of the m-PCR assay. Initially, the positive control strain B1644 (accession ID: GCA_047922575.1) was cultured on MacConkey agar, and a single colony was inoculated into Mueller–Hinton Broth, followed by adjustment to 0.5 McFarland standard. The suspension was serially diluted in sterile saline from 10^−1^ to 10^−8^, and 10 µl from each dilution was plated on MacConkey agar to determine the c.f.u. per millilitre after 24 h incubation at 37 °C. Additionally, 200 µl from each dilution was used for DNA extraction, followed by m-PCR amplification to detect target genes. The experiment, performed in duplicate, determined sensitivity by consistently identifying the lowest dilution (c.f.u. per millilitre).
These DNA samples served as templates for PCR amplification. Further, the DNA was serially diluted to achieve concentrations ranging approximately from 50 ng l^−1^ to 70 pg l^−1^. Subsequently, 2 µl of each dilution was employed as a template to evaluate the sensitivity.
Results
Screening of gene targets
A total of 12,846 genomes carrying at least 1 hypervirulence-associated marker (iucA, rmpA, rmpA2, iroB and peg-344) were identified through a comprehensive screening of the global collection in the NCBI Pathogen Detection Database Supplementary Material 1 (Table S3) and were analysed using Kleborate. However, in line with current consensus definitions, only 2,641 genomes that harboured all 5 core hvKp markers (iucA, rmpA, rmpA2, iroB and peg-344) were classified as hvKp. Among these, 576 (21.8%) co-harboured carbapenemase genes, representing CR-hvKp. Notably, 1,790 of these hypervirulent isolates lacked both ESBL and carbapenemase genes. Despite the absence of carbapenemase and traditional ESBL genes in a subset of isolates, blaCTX-M was identified in 60.38% (7,757 out of 12,846) genomes (indicating a high underlying ESBL burden in the global dataset). In the virulence marker-positive cKp group, the iucA gene was the most prevalent, detected in 96.32% (12,374 out of 12,846) of genomes, followed by peg-344 in 73.9% (9,495 out of 12,846), rmpA2 in 70% (8,992 out of 12,846), rmpADC in 48.25% (6,199 out of 12,846) and iroB in 30.83% (3,961 out of 12,846). Additionally, 68.72% (8,828 out of 12,846) of the genomes carried carbapenemase genes. Among these, blaKPC variants were the most common (4,253 out of 8,828), followed by blaNDM (2,652 out of 8,828) and blaOXA-48-like (2,429 out of 8,828).
A subset analysis of genomes from Indian clinical settings revealed a similar trend in virulence determinants but notable differences in resistance profiles. In this dataset, iucA was detected in 99.6% (523 out of 525) of genomes, while rmpADC was in 16% (87 out of 525), rmpA2 was present in 36% (189 out of 525), iroB in 16% (84 out of 525) and peg-344 in 38.09% (200 out of 525). However, the resistance landscape was as follows: 69.71% (366 out of 525) were ESBL producers carrying blaCTX-M, 76.95% (404 out of 525) of Indian genomes carried carbapenemase genes, predominantly blaOXA-48-like (93.81%, 379 out of 404), followed by blaNDM (10.89%, n=44/404), with some percentage among them having both the genes, and 4 isolates carrying blaNDM co-carried blaKPC gene and were detected.
Of the 525 Indian genomes analysed, 13.1% (69 out of 525) carried all 5 virulence genes. Among these, 15.9% (11 out of 69) also harboured carbapenemase genes, representing CR-hvKp. The remaining 81.2% (56 out of 69) lacked both ESBL and carbapenemase genes, suggesting that a majority of hypervirulent Kp in this setting circulate in non-ESBL, non-carbapenemase backgrounds.
Primer design and in silico validation
We selected five candidate genes (rmpA, rmpA2, iucA blaNDM and blaOXA-48-like) to differentiate CR-hvKp. These targets were selected based on their prevalence patterns observed in both global and Indian datasets. Primers for iroB, peg-344 and blaCTX-M were designed but not included in the primary panel, as these genes can be used in the panel on specific requirements. Similarly, blaKPC is rarely found in Indian isolates; primers were included only for standardization purposes. All primers were designed or modified to target conserved regions of hypervirulence-associated genes (rmpA, rmpA2, iucA, iroB and peg-344) and resistance genes (blaCTX-M, blaNDM, blaOXA-48-like and blaKPC variants). Designed primers were optimized to have a G+C content near 50 mol%, a ΔTm<2 °C and a length of 18–22 bases. Evaluations using in silico PCR confirmed high specificity for target sequences and no significant off-target amplification. The designed primers generated in silico amplification products of rmpA (530 bp), rmpA2 (470 bp) and iucA (170 bp), iroB (219 bp) and peg-344 (313 bp) in hypervirulent reference strains such as K. pneumoniae KCTC 2242 (NC_017540) and K. pneumoniae NTUH-K2044 (NC_012731). The primers for blaNDM, blaCTX-M, blaOXA-48-like and blaKPC produced amplicons of 401, 560, 630 and 798 bp, respectively. The detailed list of all primers is mentioned in Table 1.
Bacterial strain and phenotypic characterization
In this study, 150 clinical isolates Supplementary Material 1 (Table S4) of Kp associated with bacteraemia or respiratory infections were randomly selected for primer evaluation and phylogenomic analysis. Following confirmation of these isolates as Kp, they were assessed for hmv using the string test, which identified 22% (33 out of 150) as hmv-positive. Additionally, the isolates exhibited significant AMR, with only 30% (45 out of 150) showing susceptibility to carbapenems, specifically ertapenem and meropenem.
Genome-based population structure of bacterial strains
The MST of the Kp study isolates reveals a diverse population structure, with clear clustering based on sequence types (STs). The predominant clone was ST2096, which comprised 26.7% (40 out of 150) isolates, indicating its dominance within the studied population. ST147, the second largest cluster with 21.3% (32 out of 150) isolates, is a globally recognized high-risk clone associated with carbapenem resistance. Another prominent cluster, ST231, associated with 13.3% of isolates, is notable for its strong association with AMR genes in Indian clinical settings. Hypervirulent clones, including ST23 and ST65, were observed in smaller clusters with 4% (6 out of 150) and 1.3% (2 out of 150) isolates, respectively. Additionally, the population demonstrates considerable genetic diversity, with smaller clusters of STs such as ST101, ST14, ST45 and ST16. The phylogenetic relationships inferred from the MST highlight the coexistence of MDR and hypervirulent clones, with evidence of convergence in certain lineages. Based on Kleborate, among the 150 clinical isolates, 105 were CRKp, and 45 were non-CRKp. Of the CRKp group, only two isolates (1.9%) carried all five virulence genes (rmpA, rmpA2, iucA, iroB and peg-344) and were classified as CR-hvKp. The majority, 79 isolates (75.2%), had less than 5 virulence genes (typically iucA) and were grouped under the virulence marker-positive cKp, while 24 (22.9 %) had no virulence genes. Among the 45 non-CRKp isolates, 22 (48.9%) were classified as hvKp (all 5 virulence genes), 12 (26.7 %) as cKp with partial virulence and 11 (24.4%) lacked all virulence genes Supplementary Material 2 (Table S1).
The MST based on cgMLST revealed the clonal distribution of Kp isolates. The predominant STs were ST2096 (40 isolates), ST147 (32 isolates) and ST231 (20 isolates), forming major clusters that were centrally positioned, linking multiple other STs and suggesting their epidemiological significance. Other notable STs included ST23, ST15, ST16, ST395 and ST420 (5–6 isolates each) (Fig. 1).
MST for n=150 isolates based on cgMLST. The tree was generated using GrapeTree, with nodes representing different STs. Node sizes are proportional to the number of isolates within each ST, and colours differentiate STs. The scale represents the genetic distance.
Optimization of PCR conditions
The m-PCR assay was optimized to detect five target genes (iucA, bla_NDM_, rmpA, rmpA2 and blaOXA-48-like) through a stepwise approach, progressing from a duplex to a pentaplex assay. The optimal annealing temperature was determined to be 50 °C, with 35 cycles ensuring successful amplification. This condition provided consistent and reliable results for all target genes when multiplexed Supplementary Material 2 (Fig. S3). For all the optimization experiments, positive control B1644 (accession ID: GCA_047922575.1) was used.
Additionally, markers such as iroB, peg-344 and blaKPC were optimized as an extended version of the multiplex assay. The detailed cyclic conditions for all the markers are mentioned in Table 2. All the target genes were also amplified in singleplex PCR, confirming the functionality of individual primers. The m-PCR assay also demonstrated successful amplification of the selected markers shown in Fig. S4 (Supplementary Material 2). Agarose gel electrophoresis confirmed the successful amplification of target genes in the m-PCR assay. Various combinations of genes were tested to ensure optimal amplification without cross-reactivity (Fig. S5). Distinct bands correspond to iucA, rmpA, rmpA2, iucA, peg-344, blaNDM, blaOXA-48-like and blaCTX-M-15, all observed in a single MDR-hvKp clinical isolate (ref ID:GCA_047922635.1), validating the specificity of the assay as observed in Fig. 2.
m-PCR detection of resistance and virulence genes in Kp. Lanes show amplification of rmpA (530 bp), rmpA2 (470 bp), iucA (170 bp), iroB (219 bp), peg-344 (313 bp), blaNDM (401 bp), blaOXA-48-like (630 bp) and blaCTX-M-15 (560 bp) genes in 1 single Kp isolate, which is MDR-hvKp. A 100 bp DNA ladder is used. The presence of multiple bands indicates convergence of resistance and virulence traits.
Sensitivity and specificity
The assay demonstrated 100% specificity, with no amplification observed in non-target bacterial strains. Sensitivity tests revealed a consistent detection limit of 1 ng µl^−1^ for the target genes across triplicate experiments Supplementary Material 2 (Figs S1 and S2). Amplification results confirmed the presence of virulence genes (rmpA, rmpA2 and iucA) and resistance genes (blaNDM and blaOXA-48-like), with successful detection up to a bacterial load corresponding to a 10⁻² dilution (525 c.f.u. ml^−1^). For standardization, positive control B1644 (accession ID: GCA_047922575.1) was used.
To assess the feasibility of detecting multiple targets in a single reaction, we tested different gene combinations in m-PCR assays. Various sets of hypervirulence (rmpA, rmpA2, iucA, iroB and peg-344) and resistance markers (blaNDM, blaOXA-48-like, bla_KPC_ and blaCTX-M) were evaluated to determine compatibility and amplification efficiency. The optimized pentaplex assay (iucA, blaNDM, rmpA, rmpA2 and blaOXA-48-like) consistently yielded distinct and reproducible bands. Additional genes, including iroB, peg-344, blaKPC and blaCTX-M, were incorporated in extended multiplex reactions, demonstrating successful amplification without cross-reactivity. These results confirm the adaptability of the assay for targeted applications, allowing for tailored detection based on specific diagnostic needs, as shown in Fig. S6 (Supplementary Material 2) . The blaCTX-M, although not included in the primary multiplex panel, was standardized using the same cycling conditions and showed amplification when run alongside virulence markers, as shown in Fig. S7 (Supplementary Material 2).
Phenotypic and PCR correlation of hypervirulence and carbapenem resistance
The correlation between the hmv phenotype and virulence-associated markers was evaluated to assess the diagnostic accuracy of the string test. Among 150 isolates, the string test identified hmv in 33, of which 79% (26 out of 33) carried at least 1 virulence-associated gene (rmpA, rmpA2 and iucA). However, 76% (88 out of 116) of hmv-negative isolates also harboured 1 or more of these markers. These findings indicate that the string test does not reliably identify hvkp or correlate with any pathotype as per the hvkp definition. In contrast, resistance phenotypes showed perfect correlation with genotypic findings: all 105 phenotypically carbapenem-resistant isolates carried either blaOXA-48-like (59 out of 105), blaNDM (21 out of 105) or both genes (25 out of 105), while none of the carbapenem-susceptible strains tested positive for these genes by PCR.
WGS and PCR correlation of hypervirulence and carbapenem resistance
WGS and m-PCR analysis demonstrated a 100% correlation in detecting hypervirulence and carbapenem resistance determinants. All isolates carrying key virulence genes (iucA, rmpA, rmpA2, *iroB *and peg-344) in WGS were also confirmed by PCR, underscoring the reliability of the selected molecular markers. Similarly, carbapenem-resistant isolates harboured blaOXA-48-like, blaNDM or both, with perfect concordance between WGS and PCR results. No discrepancies were observed, indicating that the developed m-PCR-based screening effectively detects hypervirulence and resistance markers with the same accuracy as WGS.
Discussion
Healthcare-associated infections caused by Kp remain a critical global challenge due to their high prevalence, widespread AMR and diverse virulence characteristics [19]. The increasing incidence of CRKp strains has further complicated this issue, as many of these strains carry virulence-associated genes (rmpA, rmpA2, iucA, iroB and peg-344) alongside resistance determinants, resulting in severe and often untreatable infections [1534]. Complicating their detection, many of these convergent clones, which exhibit both multidrug resistance and hypervirulence traits, lack the hmv phenotype traditionally assessed using the string test. This limitation significantly undermines the utility of phenotypic methods in accurately identifying these high-risk strains. To address these diagnostic challenges, we have attempted to systematically screen global hvKp genomes and developed a robust m-PCR assay to accurately identify CR-hvKp, addressing key gaps in existing diagnostic approaches.
Genome analysis of ~12,000 hvKp genomes from the NCBI Pathogen Detection Database revealed a high prevalence of hypervirulence-associated genes, with iucA (aerobactin) being the most frequently detected, followed by peg-344/rmpA2. While less common, other hypervirulence markers such as iroB and rmpA may also be useful depending on their regional distribution. Carbapenemase genes were identified in 68.7% (8,829 out of 12,846) of the genomes, with blaKPC being the most prevalent globally. However, analysis of Indian clinical isolates demonstrated a predominance of blaOXA-48-like genes, followed by blaNDM, highlighting significant regional differences in resistance mechanisms. These findings are consistent with previous reports indicating that blaKPC is the dominant carbapenemase globally, whereas carbapenem resistance in Kp in India is often associated with endemic clones harbouring blaOXA-48-like and blaNDM [3537]. Together, these genes serve as critical markers for both virulence and drug resistance, enabling comprehensive pathotype classification of clinical isolates. MST analysis of our 150 clinical isolates identified distinct clones circulating in India, including ST2096, ST147 and ST231, which differ from the globally dominant hospital-associated clones. Notably, 58% (87 out of 150) of the isolates were MDR Kp strains carrying at least 1 hypervirulence-associated gene, reflecting a substantial burden of strains with partial virulence potential. This high prevalence of MDR-Kp strains carrying one or more virulence markers underscores the clinical relevance of the m-PCR assay in the Indian context. Given the regional differences in clone distribution and resistome profiles, we have selected nine key genetic markers that serve as critical indicators of both virulence and drug resistance, enabling comprehensive pathotype classification of clinical isolates.
The significance of this assay lies in its ability to simultaneously detect key virulence and resistance determinants within a single reaction, thereby enhancing diagnostic efficiency. By eliminating the need for sequential testing or the resource-intensive process of WGS, this approach significantly reduces turnaround time while ensuring rapid and accurate pathogen characterization. Among the 150 clinical isolates tested, the assay demonstrated 100% concordance with WGS for iucA, rmpA, rmpA2, blaNDM and blaOXA-48-like, with a detection sensitivity of 1 ng µl^−1^ DNA or 525 c.f.u. ml^−1^. This performance far exceeds that of the string test, which identified hmv in only 33 of 150 isolates, failing to detect another 88 isolates that harboured virulence markers. These findings underscore the limitations of phenotypic screening for hypervirulence. Such limitations, consistent with the findings of Russo et al. [23] and Zhu et al. [24], emphasize the unreliability of phenotypic tests for hvKp detection, particularly in cases where mutations in rmpA/rmpA2 or alternative virulence factors like peg-344 drive pathogenicity [2738]. In contrast, all 105 carbapenem-resistant isolates exhibited perfect phenotypic–genotypic correlation, reaffirming the assay’s utility for CRKp detection. The ability to rapidly and accurately identify CR-hvKp could significantly reduce mortality rates, which often exceed 50% in delayed-treatment cases [15], and enhance antimicrobial stewardship, particularly in high-prevalence settings such as India, where 57% of Kp bloodstream infections are carbapenem-resistant [10].
Previous studies, such as those by Yu et al., have attempted to differentiate carbapenem-resistant from hypervirulent Kp strains using m-PCR assays [39]. While they developed three sets of multiplex primers wherein the first set of primers was designed specifically for certain STs (ST11/258, ST23, ST86, ST65 and ST375), the second set was for capsular polymerase genes specific to various K types like K1, K2, KL64 and KL47 [39]. Similarly, another study developed an m-PCR assay targeting seven virulence genes (rmpA, allS, kfu, iuc, iro, fimH and uge) along with K1/K2 capsular serotypes, enhancing the detection of hypervirulent strains [40]. More recently, LAMP and multiplex qRT-PCR assays offer improved sensitivity and specificity for detecting hypervirulent Kp [41]. While these assays contribute to rapid and specific identification, the m-PCR approach is confined to predefined virulence genes and capsular types, potentially overlooking emerging genetic variants and converging traits. A previous study from our setting [26] prioritized iucA as the sole marker for hvKp detection. However, our inclusion of multiple virulence markers addresses variability in rmpA/rmpA2, as highlighted by Lin et al. [27]. Our assay, in contrast, provides a more comprehensive solution by simultaneously detecting both virulence and resistance factors, overcoming the limitation of focusing on a narrow set of markers. Our assay is designed to work effectively within the Indian subcontinent’s genomic diversity, including prevalent STs like ST2096, ST147 and ST231, which are not commonly identified in studies from regions such as the USA. This region-specific focus is essential for the accurate diagnosis of Kp infections and underscores the importance of tailored diagnostic tools.
A key strength of our m-PCR assay is its comprehensive detection capability, enabling the simultaneous identification of all three major Kp pathotypes (CRKp, hvKp and CR-hvKp) along with the virulence marker-positive cKp. Unlike the string test, which is subjective and prone to false negatives, this molecular approach accurately detects both virulence and resistance determinants, ensuring earlier diagnosis and intervention critical for preventing severe hvKp infections. Additionally, the assay’s high specificity ensures that only Kp genes are amplified, eliminating the risk of cross-reactivity with other bacterial species. To further enhance its clinical utility, we have incorporated regionally relevant targets, optimizing five core genes (iucA, rmpA, rmpA2, blaNDM and blaOXA-48-like) while also allowing for the inclusion of peg-344, iroB, blaKPC and blaCTX-M based on epidemiological needs. These adaptations improve the global applicability of the assay. To better scope and contextualize the performance of our m-PCR assay, we compared it with several recently published assays in Table S2 Supplementary Material 2 [4243]. Compared to WGS and singleplex PCR, which are expensive and time-consuming, this m-PCR assay offers a cost-effective, high-throughput solution suitable for routine diagnostics, particularly in resource-limited settings. Since this assay has been validated on diverse clinical isolates across India, it offers a high-throughput, reliable alternative to traditional diagnostic methods, making it especially valuable for clinical diagnostics in Indian hospitals and similar global settings. Despite these strengths, there are limitations to the m-PCR assay. We have demonstrated strong concordance with WGS-based data; we did not conduct a head-to-head comparison with other amplification platforms such as qPCR or LAMP; this remains a valuable avenue for future benchmarking. While blaCTX-M was not multiplexed with the core five-gene panel, it was successfully optimized under the same cycling conditions and can be included separately. This design preserves assay flexibility and enables regional customization based on resistance profiles. Our current validation cohort was cross-sectional, and the performance of this assay in longitudinal surveillance or outbreak settings remains an important future direction to assess its real-time epidemiological utility. Despite the fact that the isolates originated from distinct patients, we cannot completely exclude the possibility that clonal spread could occur within the same ward or hospital. This could help clarify the significant genetic similarity noted and should be taken into account when analysing prevalence and the distribution of resistance and virulence. While the assay targets the most informative virulence and resistance genes relevant to CR-hvKp detection, additional modules such as capsular typing (K-locus) and virulence genes associated with biofilm formation could be explored to expand diagnostic depth.
In conclusion, this study presents the development and validation of an m-PCR assay that offers a significant improvement over existing methods for detecting Kp pathotypes in clinical isolates. By simultaneously detecting virulence and resistance genes, the assay provides a comprehensive diagnostic tool that is suitable for rapid clinical implementation. Beyond clinical diagnostics, this assay enhances molecular surveillance and outbreak investigations by enabling real-time tracking of strain prevalence. Its ability to rapidly and accurately identify carbapenem-resistant and hypervirulent Kp supports infection control efforts, targeted interventions and antimicrobial stewardship in both healthcare and community settings. Overall, this assay provides a valuable solution for managing Kp infections in India and similar high-burden regions, addressing the growing challenge of AMR and hypervirulence through efficient and accessible molecular diagnostics.
Supplementary material
10.1099/jmm.0.002090Supplementary Material 1.
10.1099/jmm.0.002090Supplementary Material 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shon AS Bajwa RPS Russo TA Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed Virulence 2013410711810.4161/viru.2271823302790 PMC 3654609 · doi ↗ · pubmed ↗
- 2Catalán-Nájera JC Garza-Ramos U Barrios-Camacho H Hypervirulence and hypermucoviscosity: two different but complementary Klebsiella spp. phenotypes?Virulence 201781111112310.1080/21505594.2017.131741228402698 PMC 5711391 · doi ↗ · pubmed ↗
- 3Gorrie CL Mirceta M Wick RR Edwards DJ Thomson NR et al Gastrointestinal carriage is a major reservoir of Klebsiella pneumoniae infection in intensive care patients Clin Infect Dis 20176520821510.1093/cid/cix 27028369261 PMC 5850561 · doi ↗ · pubmed ↗
- 4Martin RM Cao J Brisse S Passet V Wu W et al Molecular epidemiology of colonizing and infecting isolates of Klebsiella pneumoniaem Sphere 20161 e 00261-1610.1128/m Sphere.00261-1627777984 PMC 5071533 · doi ↗ · pubmed ↗
- 5Meatherall BL Gregson D Ross T Pitout JDD Laupland KB Incidence, risk factors, and outcomes of Klebsiella pneumoniae bacteremia Am J Med 200912286687310.1016/j.amjmed.2009.03.03419699383 · doi ↗ · pubmed ↗
- 6Podschun R Ullmann U Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors Clin Microbiol Rev 19981158960310.1128/CMR.11.4.5899767057 PMC 88898 · doi ↗ · pubmed ↗
- 7Russo TA Marr CM Hypervirulent Klebsiella pneumoniae Clin Microbiol Rev 201932 e 00001-1910.1128/CMR.00001-1931092506 PMC 6589860 · doi ↗ · pubmed ↗
- 8Dong N Yang X Chan EWC Zhang R Chen S Klebsiella species: taxonomy, hypervirulence and multidrug resistance E Bio Medicine 20227910399810.1016/j.ebiom.2022.10399835405387 PMC 9010751 · doi ↗ · pubmed ↗