Antiviral candidates for enterovirus 71: targeting viral proteome and stage-specific lifecycle interventions
Jiayi Chen, Tianqi Zhao, Min Ji, Binghui Xia

TL;DR
This paper explores potential antiviral treatments for Enterovirus 71 by targeting its life cycle and proteome.
Contribution
The paper identifies new drug development strategies by focusing on virus-host interactions during the EV71 life cycle.
Findings
EV71 infections lack approved antivirals or vaccines.
Interfering with virus-host interactions offers multiple antiviral targets.
Alternative drug development avenues are proposed alongside known antiviral candidates.
Abstract
Enterovirus 71 (EV71) is a pathogen of concern, especially after its reemergence in most parts of Asia. This virus can lead to severe neurological complications and death, particularly in infants. There are no approved antivirals to prevent or treat EV71 infections, or valid vaccines. EV71 relies on precise virus-host interactions that take place during the viral life cycle, and interference with these offers numerous targets for antiviral strategies that are explored herein. We highlight known antiviral candidates and also alternative avenues to develop novel drugs.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
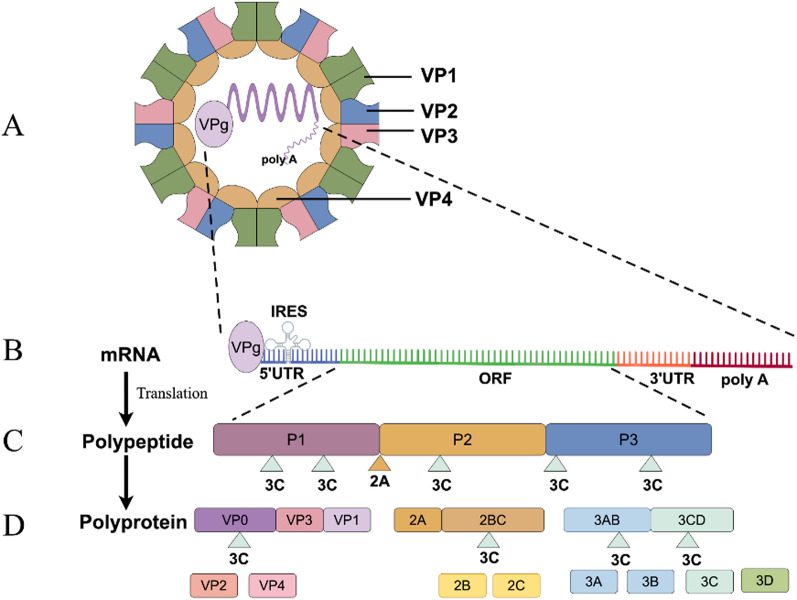 Figure 1
Figure 1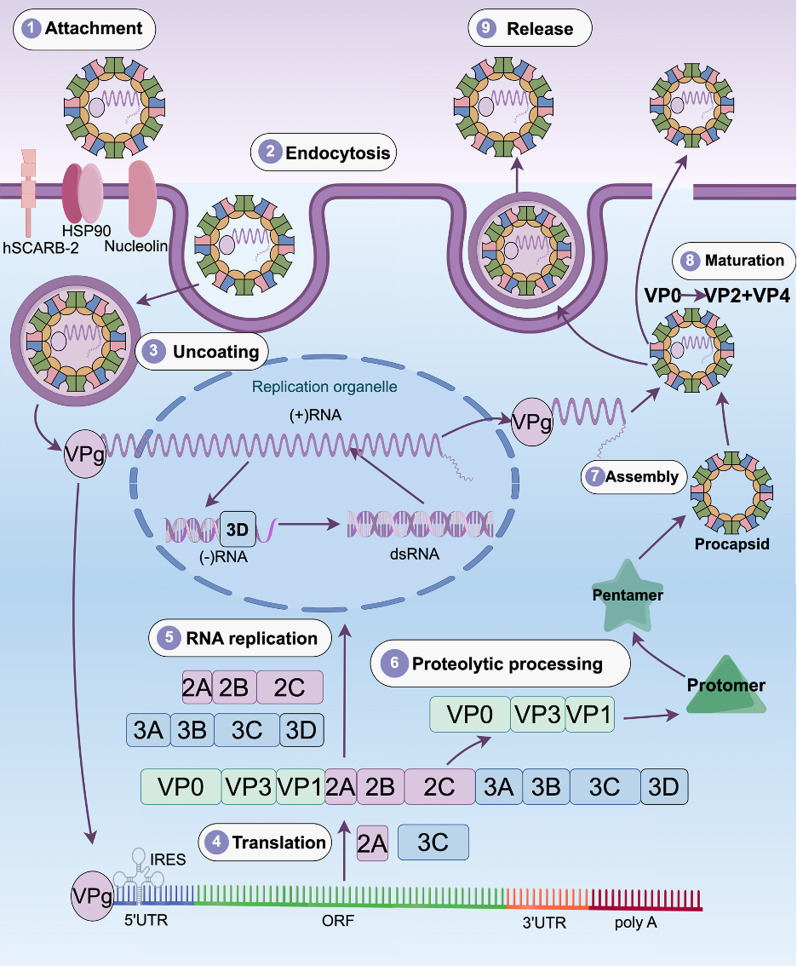 Figure 2
Figure 2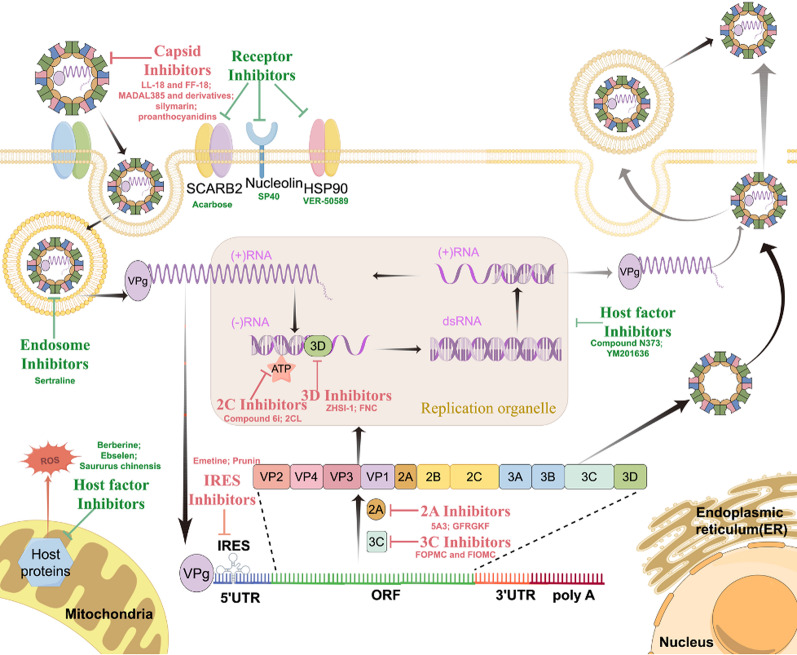 Figure 3
Figure 3- —Naval Medical University Innovation Ability Training Program
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsViral Infections and Immunology Research · Viral gastroenteritis research and epidemiology · interferon and immune responses
Introduction
Enteroviruses belong to the Enterovirus genus in the family Picornaviridae [1] and cause several clinical disorders that include neurological diseases. In the genus Enterovirus, enterovirus 71 (EV71) is one of the serotypes in the enterovirus A species (EV-A) [2] that only infects humans. EV71 is further divided into eight genotypes (A to H); B and C have five subtypes each (B1-B5 and C1-C5, respectively), where C1 and C4 are the most prevalent globally [3, 4].
Enterovirus 71 (EV71) was first described in 1974 [5] after analysis of samples collected in California over the previous four years from patients with disease of the Central Nervous System. The authors isolated the strains and presented evidence of their human origin, but the virus was circulating in the Netherlands as early as 1963 [6]. Infection by enterovirus A71 (EV71) can be asymptomatic, or it can result in a mild influenza-like illness. However, EV71 is responsible for major outbreaks of hand, foot, and mouth disease (HFMD) affecting young children, which can result in massive hospitalizations. EV71 can also cause serious neurological problems, from aseptic meningitis to acute flaccid paralysis or encephalitis [7, 8]. Indeed, EV71 is very neurotropic and highly transmissible [7, 9]. Since the initial epidemic in California in 1969, the late twentieth century has seen outbreaks in New York, several European countries and Brazil. Over the last 25 years, there have been HFMD outbreaks also in several Asia-Pacific countries [10] with mortality that typically ranged from < 0.5 to 19% [11, 12]. The unprecedented increase in the number of EV71 infections has been attributed to genotypes B and C. Among the Asia-Pacific countries, China has had the most number of EV71-associated HFMD outbreaks, with 13.7 million HFMD cases between 2008 and 2015, and 3322 deaths [13, 14].
EV71 is transmitted predominantly via the oral-fecal route [15, 16] but also through saliva [17]. Despite this, there are no antivirals available for prevention or treatment. Three vaccines that used inactivated whole viruses have been approved in China but they do not offer broad-spectrum protection against all EV71 strains [18–20], and therefore urgent development of effective countermeasures is required.
EV71 has a positive-sense, single-stranded RNA genome encapsidated in a non-enveloped icosahedral virion of about 25 nm in diameter (Fig. 1A). The viral genome (~ 7500 bases long) has a single open reading frame (ORF) flanked by conserved 5’ and 3’ untranslated regions (UTRs) (Fig. 1B). These UTRs have a covalently bound genome-linked viral protein (VPg) (at the 5’ end) and a poly A tail (at the 3’ end) [21]. The 5’ untranslated region (UTRs) of the genome (5’UTR) in EV71 is very conserved, and contains an internal ribosome entry site (IRES) that recruits ribosomes internally, driving the synthesis of the proteome [22]. The central ORF encodes a large polyprotein which can be subdivided into three precursors (P1 to P3) (Fig. 1C). The polyprotein is cleaved by viral proteases into four structural proteins (VP1-VP4, derived from P1), nonstructural proteins 2 A-2 C (derived from P2) and nonstructural proteins 3 A-3D (derived from P3) (Fig. 1D) [23]. See details in Table 1.
Fig. 1. The structure of Enterovirus 71
The non-enveloped, icosahedral EV71 viral particle has a diameter of ~ 20–30 nm and contains a single positive-sense RNA genome of about 7.4 kb. The EV71 capsid consists of four viral proteins (VP1-VP4), with VP1, VP2, and VP3 exposed on the surface and VP4 embedded inside. The genome features a central open reading frame, conserved non-coding regions at both ends, a covalently bound small protein virus genome-linked protein (VPg) at the 5’ end, and a poly A tail at the 3’ end. The RNA coding region is divided into three subregions: P1, P2, and P3. The P1 region encodes four viral structural proteins (VP1-VP4), while the P2 and P3 regions encode seven nonstructural proteins (2 A-2 C, 3 A-3D).
Table 1. The main characteristics and functions of structural and non-structural proteins of EV71CategoriesProteinApproximate number of amino acidsCharacteristicsFunctionsStructural ProteinsVP1297• The most exposed protein in the capsid• Contains the canyon and pocket factor• Important for receptor binding, viral entry and virion assembly [24]• Important determinant of EV71 immunogenicity and virulence [24, 25]VP2254Exposed on the surface of the viral capsidContains antibody recognition sites [26]VP3245Exposed on the surface of the viral capsidContains epitopes for neutralising antibodies [27]VP469Embedded inside the capsidEpitope reguired for viral infection and confers stability to the capsid [28]Nonstructural Proteins2 A150Has a proteinase active site• Hydrolyzes the peptide bond between P1 and P2 [29]• Inhibits the expression of host cell protein and induces cell apoptosis [30]2B99Consists two transmembrane helical regions HR1 and HR2Directly images the mitochondrial membrane potential and induces apoptosis [31]2 C329Contains ATPase activityImportant for cellular membrane rearrangement and genome replication [32]3 A87Has a C-terminal transmembrane helix which is responsible for membrane associationImportant for viral replication [33]3B22A primer to initiate RNA synthesis of 3D^pol^Important for viral replication [34]3 C183A multifunctional proteaseCuts off the connecting site of P2-P3, degrades DNA repair enzyme, and induces host cell apoptosis [35]3D462An RNA dependent RNA polymerase (RdRp) with a nuclear localization signalResponsible for RNA genome replication [36]
Protein components of EV71
Structural proteins
The virus capsid comprises 60 identical subunits (protomers) arranged as 12 pentamers, where each protomer has one copy of the four structural viral proteins VP1-VP4 [37]; VP1, VP2 and VP3 are exposed on the surface whereas VP4 is facing the inside of the capsid [38]. Thus, VP1-VP3 are the primary antigenic determinants of the capsid, and since the center of the pentamer is formed by five protruding VP1 proteins, these represent the main antigen and determine the serotype [39]. In enteroviruses, VP1, VP2 and VP3 have β-sandwich “jelly-roll” folds [38, 40] and form a depression around an icosahedral five-fold axis called the “canyon” [41], where receptors that have an immunoglobulin-like fold bind to [42].
Nonstructural proteins
P2-derived proteins. The non-structural proteins originate from precursors P2 (2 A-2 C) and P3 (3 A-3D) (Fig. 1C-D). The EV71 2 A (150 residues) is a cysteine protease (2A^pro^) with a chymotrypsin-like fold with an active site that consists of a catalytic triad formed by C110A, H21 and D39 [30, 43]. 2A^pro^ mediates the initial step of polyprotein processing, cleaving between the C-terminus of VP1 and its own N-terminus (‘2A’ in Fig. 1C) [44, 45], and hijacks host cell gene expression by cleaving eukaryotic initiation factor 4G (eIF4G) [30], whereas 3C^pro^ (see below) performs subsequent polyprotein proteolysis [35].
2B is a membrane protein of 99 amino acids with two transmembrane helical domains that form ion channels critical for virion release [31] and may release calcium into the cytoplasm inducing apoptosis [46].
Lastly, 2 C protein mainly functions as an NTPase, where ATP hydrolysis helps unwind RNA. The crystal structure [32] contains an adenosine triphosphatase (ATPase) domain that belongs to SF3 helicases of the AAA + ATPase superfamily, a cysteine-rich zinc finger and a C-terminal α-helix involved in homo-oligomerization via interaction with a deep pocket formed between the ATPase and the zinc finger domains of a neighboring monomer. Thus, antivirals may be targeting its enzymatic activity and its reliance on self-oligomerization. Additionally, the N-terminal amino acids (~ 50 residues) have potential membrane-binding activity and are involved in membrane remodeling and in the formation of the viral replication complex, where 2 C associates with host reticulon 3 and with the viral double-stranded RNA [47].
P3-derived proteins. The 3 A protein (87 amino acids long) also has a C-terminal transmembrane helix which is responsible for membrane association [48], whereas 3B is a ~ 20 residues long peptide known as VPg (virus genome-linked protein) (Fig. 1B) which also binds at the bottom of the palm domain of the 3D^pol^ molecule (see below) [34]. 3 A, together with the host phosphatidylinositol 4-kinase IIIβ (PI4KB), acyl-coenzyme A binding domain containing 3 (ACBD3) and oxysterol-binding protein (OSBP) are involved in the formation of replication organelles [49], which requires the hijacking of host lipid homeostasis. Indeed, 3 A recruits PI4KB via the Golgi-resident ACBD3 protein, creating a phosphatidylinositol 4-phosphate (PI4P)-rich microenvironment, in turn recruiting 3D^pol^ to replication sites where it catalyzes the synthesis of RNA [50]. The structure of this ternary complex is still unresolved, but some structural data of individual components and their interactions are available. For instance, low resolution SAXS-based hybrid structures of PI4KB and ACBD3 and their association have been reported [51]. Furthermore, the structure of the complex formed by ACBD3 GOLD domain and enteroviral 3 A proteins has been determined, e.g., in EV71 3 A (PDB ID: 6HLW), revealing critical interaction motifs [52]. Additionally, PI4KB and ACBD3 have been solved by solution NMR (PDB ID: 2N73), with insights into their interface [53].
The availability of these data suggests that this complex may be targeted for antiviral development. Interestingly, compound N373, which selectively inhibits PI4KB, resulted in resistant mutants localized in protein 3 A [54], suggesting that PI4KB plays a role in the interaction or replication mechanism involving protein 3 A. This is a challenge for identifying drug binding sites in the viral proteome by the method of obtaining revertant mutants; these mutants may appear in viral proteins even though the actual target is a host protein. Therefore, when developing antiviral drugs, it is essential to consider the interplay between viral and host proteins to ensure efficacy and specificity.
The precursor fusion protein 3CD (Fig. 1D) degrades DNA repair enzymes and induces host cell apoptosis [35] and most importantly, leads to formation of 3C^pro^ and 3D^pol^. Similar to other picornaviruses, EV71 3C^pro^ (183 residues) forms a chymoprotease-like fold with two topologically equivalent antiparallel six-stranded β-barrel domains [55]. Located between the two, there is a long shallow groove for substrate binding, but in EV71 this site is very exposed to the solvent. 3C^pro^ cuts itself from the P3 precursor and cuts P2 and P3 to form the final non-structural proteins (see ‘3 C’ in Fig. 1C-D) or the precursor fusion 3CD [56]. 3C^pro^ contains a surface β-ribbon loop, at the base of which Gly123 and His133 form a flexible hinge important in the proteolytic activity [55]. 3C^pro^ also has RNA binding ability and is involved in replication [57].
Lastly, 3D protein (3D^pol^, 462 amino acids) is an RNA-dependent RNA polymerase (RdRp) critical for RNA replication [34]. 3D polymerase is activated by the proteolytic cleavage of the 3CD precursor protein. This activation mechanism is conserved among picornaviruses, even when the 3D protein does not start with a glycine residue [58]. Viral replication complexes are associated with host cell membranes, particularly the endoplasmic reticulum (ER). Recruitment of 3D protein to these membranes involves interactions mediated by the negative charge of the membrane and the 3AB protein [59]. The latter facilitates membrane remodeling and formation of double-membrane vesicles [60]. The 3D protein contains three domains: RNA-binding, catalytic (responsible for RNA synthesis) and inhibitory (which regulates polymerase activity) [61]. Inhibitors targeting the 3D polymerase, discussed later, represent a promising strategy for antiviral therapy, as they can effectively block viral replication [62]. Overall, the 3D protein is central to the replication cycle of EV71, making it a key target for both mechanistic studies and therapeutic interventions [63].
Because of their enzymatic activity, 2 A, 3 C and 3D proteins have been the target for several antivirals. In contrast, membrane-associated 2B, 2 C and 3 A, and intermediates 2BC and 3AB, which are involved in replicating organelle formation have a limited number of drugs reported. This is clearly limited by the lack of structural information for these membrane-associated proteins. Channel activity (2B) and membrane remodeling and oligomerization (2 C and 3 A) should be explored further as targets by using platforms such as cryo-EM or solid state NMR.
Lifecycle of EV71
Virus entry into susceptible host cells starts with attachment to the cell surface and receptor binding (see Fig. 2) to one of the several human receptors identified, e.g., the human scavenger receptor class B member 2 (SCARB2) [64], human P-selectin glycoprotein ligand-1 (PSGL-1) [65], sialic-acid-linked glycan [66], human annexin 2 protein [67] and heparan sulfate glycosaminoglycan [68]. Binding is followed by endocytosis which varies among serotypes and cell types [69]. Binding of receptors to the canyon of the capsid causes expulsion of a hydrophobic “pocket factor” (a lipid molecule) which signals genome release into the cytoplasm [70] through a proteinaceous pore spanning the endosomal membrane [71]. In EV71, this requires both receptor binding and acidification of the endosomal lumen [72, 73]. Upon cytoplasmic entry, RNA is translated into a single polyprotein, and serves as a template for genome replication by 3D^pol^. The newly generated RNA either replicates or forms new virions. After an empty procapsid is formed, RNA is enveloped within the procapsid and triggers cleavage between VP2 and VP4 (Fig. 1D), thus generating mature infectious virus particles [74]. The virion can exit the cells via cell lysis but also through subversion of the autophagic machinery (Fig. 2). Overall, the life cycle of EV71 and enteroviruses in general offer several targets for antivirals [75]. For example, picornavirus genome delivery into the cytoplasm is facilitated by a mechanism previously implicated in the clearance of intracellular bacteria. Indeed, a host lipid-modifying enzyme, adipose-specific phospholipase A2 (PLA2G16), prevents the autophagic degradation of permeated endosomes [76], and therefore PLA2G16 is a possible drug target, as are other host enzymes that regulate autophagy, which are controlled by EV71 and other enteroviruses [77, 78].
The present review focuses on novel antiviral candidates reported in approximately the last five years that target parts of the EV71 life cycle. Inhibitors with specific targets are discussed, while those with unclear targets are tabulated.
Fig. 2. Schematic overview of Enterovirus 71 lifecycle
EV71 binds to one or more cell surface receptors, triggering viral endocytosis. The virus particles then transport to endosome and release the genome through endosomal membrane holes. Upon cytoplasmic entry, RNA is translated into four structural proteins (VP1-VP4) and seven non-structural proteins (2 A-2 C, 3 A-3D). Simultaneously, RNA serves as a template for genome replication under 3D^pol^ action. The newly generated RNA either replicates or forms new virions. The structural proteins further form an empty capsid. Then RNA is enveloped within the Procapsid and generate mature infectious virus particles. Finally, progeny viruses are released.
Virus-directed antivirals
Antivirals for EV71 can be directed at the virus or at the host (Fig. 3). This section will describe those directed at the virus, which are also shown in Table 2. VP1, 2 A, 3 C and 3D are considered to be the most promising targets since their structures are known.
Fig. 3. Schematic diagram of antivirals for EV71 with different targets
Antivirals targeting viral components and host cells are presented separately with different colored arrow lines. Virus-directed antivirals are presented in red color, while host-directed antivirals are presented in green color. The most promising antiviral candidates for each category have been selected and presented in the figure.
Inhibitors targeting VP1 proteins
In picornaviruses, antivirals that target the canyon formed by VP1 can show high affinity [79]. In EV71, this canyon is shallower than that in other enteroviruses and VP1 shows high variability which can result in fast development of resistance. However, highly conserved residues, especially Arg and Pro, have been identified at the intersection between the loops and the strands in the β-barrel in VP1 sequences from all enteroviruses [80, 81]. Targeting these should not only slow down resistance but also lead to pan-enteroviral inhibitors. In general, the capsid has been targeted by peptides, synthetic compounds or natural compounds.
Peptides
Although therapeutic peptides have poor in vivo stability and are attacked by enzymes, they have lower off-target toxicity and lower emergence of resistance compared to small drugs (see review in [82]). Historically, lactoferrin—an 80-kDa iron-binding glycoprotein—is one of the earliest EV71 inhibitors: both bovine and human forms bind the capsid protein VP1 and also cellular heparan sulfate receptors, thus inhibiting viral adsorption to cells [83, 84]. This dual binding mechanism was confirmed by ELISA-based binding assays demonstrating lactoferrin-VP1 interaction, which could be competitively blocked by anti-VP1 antibodies [84].
Since proteins and peptides have low membrane permeability, they mostly target the capsid. Other inhibitors are antimicrobial peptides (AMPs), which are part of the innate immune system [85]. For example, cathelicidin (the mature and processed form of cathelicidin in humans is LL-37) is effective against enveloped viruses, but also against non-enveloped ones [86]. In EV71, LL-37 directly inhibited viral binding to cells and inhibited EV71 replication by regulating the antiviral immune response increasing basal interferon-β (IFN-β) expression [87]. Shortening peptides results in general in better bioavailability [88]. Thus, removal of first and last five amino acids of LL-37 produced peptides LL-18 and FF-18 [89]. These 27-residue peptide analogues showed more potency than the parent peptide LL-37 and were found to directly bind EV71 virus particles at the viral canyon region, blocking virus-receptor interactions and inhibiting viral uncoating. This was confirmed by monitoring the changes in the amide region of the peptide by NMR after exposure to the virus and co-immunoprecipitation of the peptides with capsid proteins VP1-VP3. Interestingly, prevented genome release after uncoating and resistance was not observed even after multiple passages, which suggests that mutations that prevent peptide binding would also be deleterious for viral stability [90].
Peptides may also be derived from the VP1 capsid protein of EV71. For example, 95 overlapping 15-mers spanning VP1 were tested, and some inhibited EV71 [91]. One of these peptides, SP81, showed direct virus inactivation of EV71 with an IC_50_ of 8.076 µM, supporting its direct binding to the viral surface [92] although whether VP1 is the actual target remains known.
Synthetic compounds
Early efforts yielded compounds like pleconaril, which demonstrated efficacy against other enteroviruses, but activity against EV71 was poor, possibly because insufficient binding to the viral canyon [93, 94]. In contrast, vapendavir forms hydrogen bonds with VP1 residues, e.g., Asp112/Ile113, has anti-EV71 activity and is in clinical development for picornavirus infections [94].
Concurrently, significant progress has been made with novel chemotypes. Rivero-Buceta et al. [95] previously reported a family of tri-and tetrapodal Trp derivatives with prototype AL-385 that inhibited EV71 [96]. AL-385 was found to bind the 5-fold axis of the viral capsid using cryo-EM methods and showed inhibition of EV71 at nM concentrations [97]. By simplifying the scaffold, the same authors identified a second family of EV71 entry inhibitors [98], where two compounds, AL-470 and AL-471, interact with the five fold axis of the capsid (VP1 K244 and Y245). Further, SAR studies developed a new compound based on the same scaffold (a double arylated tetrapodal compound, AL-518) with EC_50_ against EV71 of 40 nM [99]. Further development has resulted in more potent multivalent Trp- and Tyr-containing fullerene hexa-adducts that target both HIV and EV71 [100]. Finally, the insertion of an amphipathic linker in a tetrapodal Trp derivative led to highly potent EV71 inhibitors in clinical isolates [101]. Although these compounds show high affinity and selectivity, and can be optimized with the availability of high resolution structures, it remains to be seen if they can be developed into non-toxic compounds in in vivo studies.
Notably, tanomastat, a repurposed synthetic inhibitor, targets VP1 by binding to its hydrophobic pocket through interactions with key residues and a conserved water molecule, thereby impeding capsid dissociation during viral entry. It exhibits broad-spectrum activity against multiple enterovirus species (A-D) and exerts multi-stage inhibition, including inhibition of viral RNA replication [102].
Natural products
Natural products can also target the capsid. A component of Ilex kaushue extracts, 3,4-dicaffeoylquinic acid (3,4-DCQA), showed broad inhibition against different EV71 genotypes by disrupting viral attachment to host cells, specifically targeting E98 and P246 in the 5-fold axis located within VP1, as shown by resistance mutants at these positions. 3,4-DCQA specifically inhibited the attachment of EV71 to the host receptor heparan sulfate (HS), but not to the scavenger receptor class B member 2 (SCARB2) and P-selectin glycoprotein ligand-1 (PSGL1) [103]. This type of interaction may be similar to that observed for the antiparasitic drug suramin (derived from azo dyes) and its sulfated derivative NF449 [104], which also binds near the 5-fold vertex and interferes with attachment to PSGL-1 and heparan sulfate, although in this case VP1 residue 244 was identified as critical for the interaction [105].
Another class of natural compounds that target VP1 are flavonoids such as silymarin or proanthocyanidins (PC). For example, silymarin could inhibit EV71 (IC_50_ of 15 µg/mL) by preventing virus attachment to host cells [106]. The mechanism was proposed to involve binding to the VP1 GH loop [107] which interacts with the uncoating receptor SCARB2 [70]. Similarly, baicalein from Scutellaria baicalensis exhibits extracellular virucidal activity against EV71 (IC_50_ = 30.88 µg/mL) by disrupting viral attachment and entry, likely through capsid-binding [106]. Proanthocyanidins (PC) may also bind VP1, and intramuscular PC therapy in EV71-infected mice improved survival rates and increased body weight [108]. However, experimental evidence of selectivity, binding sites and affinity of these compounds is lacking, which precludes further development.
Inhibitors of proteases 2Apro and 3Cpro
2 A protease
To date, 2A^pro^ remains a relatively underexplored target, with no potent 2A^pro^ inhibitors available. Early studies identified the LVLQTM peptide as a classic inhibitor of 2A^pro^, which functions as a pseudosubstrate by binding to the protease’s active site and effectively inhibits EV71 replication [109]. In recent years, research on 2 A has explored its role in immune evasion and inhibitors targeting this role may emerge as a new direction for future antiviral drug development. For example, 2A^pro^ reduces interferon-α receptor 1 (IFNAR1), antagonizing the antiviral activity of IFN-a [110]. It also inhibits the activation of Cyclic GMP-AMP Synthase-Stimulator of interferon genes (cGAS-STING) by targeting tumor necrosis factor receptor-associated factor3 (TRAF3), which helps the virus evade host immune attack [111]. Nevertheless, a region present in viral but not human proteases was identified, and peptides were selected that could bind to that region. Four peptide candidates inhibited 2 A protease activity and the crystal structure of the best peptide bound to 2 A protease was determined [112]. This inhibition may also block the cleavage of antiviral signaling proteins and interferon receptor 1. Additionally, the model peptides can be synthetically modified to resist enzymatic digestion. More recently, etoposide—a semi-synthetic podophyllum derivative—has been identified as a novel 2A^pro^ inhibitor. It targets key residues Y89 and P107 within the protease’s binding pocket, as validated by molecular dynamics simulations and mutagenesis assays. Etoposide exhibits broad-spectrum activity against EV71 genotypes A–C and CVA16, with minimal cytotoxicity even at high concentrations (100 µM). Its established clinical biosafety profile supports potential repurposing as an anti-EV71 agent [113].
3 C protease
Rupintrivir (AG-7088) is a peptide [114, 115] that binds to 3 C catalytic pocket via a covalent linkage to Cys147, although clinical trials were unsuccessful [116]. Recent efforts have developed rupintrivir analogues, such as synthetic peptide SG85 [117]. Further modifications of rupintrivir have been attempted using in silico methods [118].
A novel series of peptidomimetic aldehydes was designed to target 3 C protease of EV71 [119]. Not only the compounds showed high antiviral activity but the crystal structure of EV71 3C^pro^ in complex with compound 18p was determined at high resolution (1.2 Å), revealing a covalent linkage to catalytic Cys147 [119].
Two synthetic inhibitors of EV71 3C^pro^, propyl- and isopropyl-substituted 4-iminooxazolidin-2-one moieties (FOPMC and FIOMC) were developed from a previously reported cyanohydrin derivative. However, although these compounds showed inhibitory activity, the binding modes were only shown with in silico docking [120]. Quantitative irreversible tethering (qIT) was used to identify acrylamide fragments that target the active site cysteine of the 3 C protease (3C^pro^) of EV71, where crystal structures of fragments and enzyme were obtained [121].
Additionally, a series of 2-benzoxyl-phenylpyridine derivatives—particularly WY7 —have been identified as potent 3C^pro^ inhibitors. WY7 inhibits EV71 3C^pro^ by inserting into the substrate-binding pocket, forming hydrogen bonds with Thr142 and His161, and disrupting substrate recognition. It also moderately inhibits 3D polymerase, further enhancing antiviral efficacy with an EC_50_ of 11.29 µg/mL in RD cells [122].
Natural products and flavonoids constitute a well-characterized class of 3C^pro^ inhibitors. Quercetin, a widely distributed flavonoid, potently inhibits EV71 3C^pro^ by inserting into its substrate-binding pocket to block substrate recognition, thereby exerting antiviral activity against EV71 [123]. Luteoloside, another flavonoid, inhibits 3C^pro^ in a dose-dependent manner and acts post-attachment to reduce EV71 yields [124]. Fisetin and rutin, two additional flavonoids, directly interact with 3C^pro^ to impair its activity and suppress EV71 plaque formation [125].
Virtual screening of compounds used in traditional Chinese medicine identified salvianolic acid, a polyphenolic compound isolated from Salvia miltiorrhiza which inhibited EV71 infection [126]. However, no binding sites were experimentally determined, although in silico docking suggested binding to 3C^pro^.
D-D7 is a peptidomimetic containing 2-pyridone that originally targeted human rhinovirus 3C^pro^ [127]. This drug was repurposed to target the same enzyme in EV71 [127], thereby disrupting polyprotein processing. Future studies should optimize its pharmacokinetics, evaluate combinatorial therapies, and validate efficacy in preclinical models.
Inhibitors of 2 C protein
2 C is a helicase and an ATPase, and an attractive target for antivirals due to its high conservation [32]. Fluoxetine, a selective serotonin reuptake inhibitor (SSRI) approved by the Food and Drug Administration (FDA), only inhibits species EV B and EV D [128], but the analogue N-(4-fluorobenzyl)-N-(4-methoxyphenyl)furan-2-carboxamide could inhibit all EV strains in the sub-µM range [129]. Escape mutants were found in 2 C near the α2 helix. Using in silico methods the authors proposed a conserved binding pocket in all EVs [129].
In a different study, a quinoline formamide analogue, compound 6i, bound the ATPase domain. This drug could alleviate clinical symptoms in mice when combined with emetine, suggesting binding to 2 C although this was not experimentally validated [130].
Another promising target is the pocket formed between the zinc finger and the ATPase helicase domain, which mediates homo-oligomerization of 2 C [32]. In silico and in vitro testing for 2 C ATPase inhibition identified SJW-2 C-227 as a potent broad-spectrum compound targeting the 2 C:2 C interaction pocket [131]. Peptide 2CL, derived from the 2 C α6 helix, was designed to inhibit 2 C oligomerization by competing with the 2 C α6 helix for binding to a neighboring 2 C deep pocket, demonstrating antiviral properties against EV71 in cells, and improving the survival of mice [132].
The host ubiquitin-proteasome system can be used to degrade viral components. For example, 2 C protein is ubiquitinated by UBE3C, a host E3 ubiquitin ligase, thus restricting EV71 replication. This requires developing UBE3C activators or mimetics with potential broad-spectrum activity against enteroviruses because of 2 C high conservation. However, this strategy risks disrupting host ubiquitination, therefore future efforts should minimize cellular toxicity [133].
Inhibitors of 3Dpol
In general, 3D in picornaviruses is highly conserved and is, therefore, an attractive antiviral target [134]. Recent studies have focused on the role of 3D^pol^ beyond RdRp [134], e.g., in interaction with host proteins, where 3D^pol^ can induce cell cycle arrest, regulate host cell translation and induce apoptosis. Inhibitor ZHSI-1 significantly reduced the binding of 3D to lysosomes, inhibiting EV71 replication [135]. Therefore, targeting the lysosome-tethered Ragulator-Rag-3D complex may represent an effective therapeutic strategy. Azvudine/FNC (2’-deoxy-2’-β-fluoro-4’-azidocytidine), a novel cytidine analog originally developed for HIV treatment, binds to 3D^pol^ and incorporates into nascent RNA causing chain termination [136].
Recent advances highlight nucleotide analogs and nucleoside phosphonates as promising candidates targeting the EV71 3D protein. GS-646939, a 4’-cyano-modified C-adenosine triphosphate analog, exhibits exceptional selectivity for picornaviral RdRps, including EV71 [137]. Biochemical studies demonstrate its preferential incorporation over ATP by EV71 3D^pol^, leading to immediate chain termination via steric hindrance during translocation, even under high NTP concentrations. This mechanism contrasts with delayed termination observed in coronaviruses, underscoring its specificity for picornaviruses. In parallel, β-D-xylofuranosyl nucleoside phosphonates, exemplified by adenine derivative 1e, were designed to mimic natural nucleotides [138]. Molecular dynamics simulations suggest binding to the RdRp active site, but activity against enteroviruses is modest (EC_50_ = 16 µM for EV-68), likely due to competitive inhibition and impaired base pairing efficiency [138].
Future development of GS-646939 should focus on optimizing its broad-spectrum efficacy against emerging picornaviruses while minimizing off-target effects, potentially through prodrug engineering or combination therapies. For xylofuranosyl phosphonates, structural modifications to enhance RdRp affinity and nucleobase versatility could improve antiviral potency, particularly against EV71, which remains underexplored in current studies [137, 138]. Both classes face challenges in balancing polymerase selectivity with metabolic stability, necessitating advanced structural modeling and high-throughput screening. Collectively, these agents underscore the therapeutic potential of RdRp-targeted strategies.
Inhibitors of IRES
The IRES is a relatively new antiviral target, with inhibitors that consist predominantly of natural compounds, some of which repurposed. However, mechanisms of inhibition are poorly characterized, which prevents further development [139]. Historically, several antivirals have demonstrated IRES-targeting activity against EV71, albeit with limited translational progress. Mitoxantrone, initially identified through FRET-based screening, selectively disrupts EV71 IRES-dependent translation by binding to structural RNA motifs [140]. Similarly, the flavonoid apigenin inhibits viral translation initiation by competitively blocking interactions between the IRES and host factors hnRNP A1/A2 [141]. Lycorine indirectly impairs IRES function by arresting viral polyprotein elongation during translation [142].
Recent studies have unveiled novel IRES-targeting strategies. Notably, a binder of 5’-UTR is APOBEC3G, or A3G. This is apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G, a cytidine deaminase that inhibits the replication of several viruses. Indeed, A3G inhibited EV71 virus replication, impairing the interaction between the 5’UTR and the host protein poly(C)-binding protein 1 (PCBP1), whereas EV71 overcame A3G suppression through its non-structural protein 2 C, by inducing A3G degradation through the autophagy-lysosome pathway viruses [143]. Compound IMB-Z, a N-phenylbenzamide derivative, was found to increase the levels of intracellular A3G. This effect was unrelated to the enzymatic activity of A3G, but instead, inhibition of EV71 replication was caused by A3G interaction with viral 3D (RdRp) and viral RNA, and packaging into less infective virions. Thus induction of A3G by drugs can be used as a potential therapeutic approach to tackle EV71 infection [144].
Emetine, an antiprotozoal drug, could inhibit EV71 IRES-driven translation in a mouse model [145], whereas small molecule DMA-135 induced a conformational change in the RNA structure and stabilized a ternary complex that inhibited translation [146]. Plant compounds kaempferol or Hsp27 protein inhibitor TDP also can target IRES [147, 148], whereas prunin prevents translation [149, 150].
Table 2. Virus-directed antiviralsTarget/MechanismClassification of antiviralsName ofantiviralsStudy stagesDirection of optimizationStudy Stages/Experimental modelReferenceIn vitroIn vivoVP1Natural productMethyl linoleateThe actual target has been identified by molecular dockingIdentification of molecular mechanismsVero cells-[151]ErgosterolThe actual target has been identified by molecular dockingIdentification of molecular mechanismsVero cells-[151]Mulberroside CThe actual target has been identified by molecular dockingIdentification of molecular mechanismsRD and Vero cellsICR mice[152]Trans-2-hexenoic acid (THA)The target has been predicted by molecular dockingFurther verification of the antienterovirus spectrum with in vivo evidence and identification of molecular mechanismsHeLa cellsKunming mice[153]Rosmarinic acid (RA)The target has been predicted by molecular dockingFurther evaluation of the virucidal activity of RA in combination with other anti-EV71 inhibitorsRD cellsICR mice[154]Fangchinoline (FAN)The target has been predicted by viral resistance mutationsIdentification of the target and molecular mechanismsVero cells-[155]SilymarinThe actual target has been identified by ELISA competitive binding assay and viral resistance mutationsFurther in vivo evaluationRD cells-[107]BaicaleinThe target has been predicted by molecular docking (virucidal activity)Improvement of inhibitory activity and acceleration of virucidal effectsRD cells-[106]Proanthocyanidins (PC)The actual target has been identified by SPR assay and molecular docking analysisFurther studies in animal models and clinical researchRD and Vero cellsICR mice[108]Nucleotide sequenceDNA aptamers V7, V11, V21The actual target has been identified by enzyme linked immunosorbent assay (ELISA)Combination with nanoparticles, siRNA/miRNA, and chemical drugs to formulate new therapeutic complexesRD cells-[156]siRNA with nanoparticlesSe@PEI@siRNAThe actual target has been identified by inhibition test in vitroIdentification of molecular mechanismsSK-N-SH cells-[157]Monoclonal antibody (mAb)cD5The actual target has been identified by ELISA and Western blot assaysFurther studies in animal models and clinical researchRD cellsICR mice[158]Cathelicidin peptide analoguesLL-18, FF-18The actual target has been identified by molecular docking and inhibition test in vitroFurther studies in animal models and clinical researchRD and 293 T cellsICR mice[90]Ilex kaushue extracts3,4-dicaffeoylquinic acid (3,4-DCQA)The actual target has been identified by molecular docking and resistant virus selection experimentsIdentification of molecular mechanisms and further in vivo evaluationRD cells-[103]Tryptophan dendrimersMADAL385The actual target has been identified by resistance selection, reverse genetics and cryo-EMTo simplify and reduce the backbone of MADAL385 without affecting the antiviral activityRD cells-[101]Tyrosine (Tyr) fullerene 10The actual target has been identified by molecular dockingIdentification of molecular mechanismsRD cells-[99]AntisepticPovidone-iodine (PVP-I)The target has been predicted by Western blot analysisWhether or not PVP-I denatures other viral capsid proteins (VP2 and VP3) or viral genomeVero, RD, human primary oral fibroblast (HPOF) cells and L929 mouse cells (L-hSCARB2 cells)-[159]Small moleculeCompound 5 h, 6cThe actual target has been identified by preliminary docking assaysEvaluation of pharmacokinetic parameters and spectrum of activityRD and MRC-5 cells-[160]TanomastatThe actual target has been identified by molecular docking and live virus particle trackingFurther in vivo evaluation and clinical trialsRD and Vero cellsBALB/c mice[102]VP3ChemokinePlatelet factor 4 (PF4) or the CXC chemokine CXCL4The actual target has been identified by coimmunoprecipitation assay (IP)Identification of molecular mechanismsHEK293T and RD cellsMice[161]2 AMurine mAb5A3The actual target has been identified by indirect fluorescent assay, Western blotting and ELISAFurther studies in animal models and clinical researchVero cellsBALB/c mice[162]PeptideGFRGKFThe actual target has been identified by crystallographyFurther studies in animal models and clinical researchMolecular dynamics simulations-[112]Natural productEtoposideThe actual target has been identified by molecular docking, molecular dynamics simulations, and site-directed mutagenesisFurther studies in animal models and clinical researchRD, HEK-293T, and Vero cells-[113]2 CPyrazolopyridine-containing small moleculesCompound 7 d, 7 h, 10a, and 19The actual target has been identified by serial viral passage experiments, coupled with reverse genetics and thermal shift binding assaysThe high-resolution X-ray crystal structure of the 2 C proteinRD, A172 and SH-SY5Y cells-[163]Fluoxetine-derived small moleculesCompound 12b (and analogues 12a, 19b, 19 d)The actual target has been identified by resistance mutations and thermal shift assaysStructural optimization for enhanced potency/spectrum, mechanistic elucidation via 2 C-inhibitor complex determination, and preclinical safety/efficacy profilingHeLa, HAPI and BGM cells-[129]Quinoline formamide analogueCompound 6iThe actual target has been identified by investigating the dsRNA unwinding in cellsExtensive pre-clinical investigation as a lead compoundRD and Vero cellsKunming mice[130]Small moleculeSJW-2 C-227The actual target has been identified by differential scanning fluorimetry (DSF)To determine a structure of the compound bound to 2 CRD, HeLa and Vero cells-[131]Quinoline compoundCompound 6awThe actual target has been identified by thermal shift assay (TSA)Further studies in animal models and clinical researchRD cells-[164]Peptide2CLThe actual target has been identified by molecular dockingTo enhance the antiviral activity, stability, and bioavailabilityRD, Vero, Huh7, and 293 T cellsICR mice[132]E3 ubiquitin ligaseUbiquitin protein ligase E3C (UBE3C)The actual target has been identified by Co-IP combined with ubiquitination modification mass spectrometryThe exact combination modeVero, human embryonic kidney (HEK293T) cells and human colon cancer (HCT-8) cells-[133]3 CMacrocyclic peptidomimeticCompound 4The actual target has been identified by the crystallographic characterizationImprovements in potency and stabilityRD cells-[165]Acidic polysaccharidesLJ04The actual target has been identified by inhibition test in vitroFurther investigation of safety and efficacyMA104 and HM1900 cells-[166]Michael acceptorSLQ-4, SLQ-5The actual target has been identified by molecular dockingCrystallographic study to disclose the precise interactionRD, 293 T and Vero cells-[167]Peptidomimetic compoundRupintrivir (AG-7088)The actual target has been identified by crystallographyBetter water solubility, oral bioavailability and potent activityRD cells-[168]Synthetic peptide SG85The actual target has been identified by X-ray crystallographyImprovement of stability, bioavailability, and activityRD cells-[169]Compound 18pThe actual target has been identified by crystallographyFurther studies in animal models and clinical researchRD and Vero cells-[119]FOPMC and FIOMCThe actual target has been identified by molecular docking and molecular dynamics simulationsFurther studies in animal models and clinical researchRD cells-[120]Amino acid fragmentAcrylamide fragmentsThe actual target has been identified by quantitative irreversible tethering (qIT)Improvement of specificity and safetyQuantitative irreversible tethering (qIT)-[121]Small moleculeWY7The actual target has been identified by inhibition test in vitro and molecular dockingImprovement of inhibitory activity and further in vivo evaluation.RD cells-[122]2-pyridone-containing human rhinovirus 3 C protease inhibitorD-D7The target has been predicted by crystal structure analysisPharmacokinetics and pharmacodynamics of the compounds in animal modelsRD cells-[127]Natural productHesperidinThe actual target has been identified by molecular dockingFurther in vitro studyMolecular docking-[170]Salvianolic acid A (SA)The actual target has been identified by molecular mechanics generalized born surface area (MMGBSA) and steered molecular dynamics (SMD)To increase the contentRD cells-[126]3DPyrimidine analogsGemcitabineThe actual target has been identified by RT-PCR and western blotFurther in vivo evaluationRD cellsBALB/c mice[171]LY2334737Be used as a gemcitabine prodrug generated by linking gemcitabine covalently to a valproic acid groupSimilar dosage as clinical trialsRD cellsBALB/c mice[171]SofosbuvirAn FDA approved nucleotide analog that already in use for the treatment of hepatitis C infectionsTo test drug doses to determine the optimum dose suitable for potential clinical trialsRD cellsBALB/c mice[171]Monophosphoramidate adenosine analog prodrugRemdesivir (GS-5734)The actual target has not yet been identifiedDevelopment of a suitable animal model and combination with other virus entry inhibitorsHeLa cells-[172]Small moleculeZHSI-1The actual target has been identified by biotin tagThe precise mechanism by which ZHSI-1 interacts with 3D polymerase to modulate its functionRD, SK-N-SH, U251 and 293 T cellsC57BL/6J (B6) mice and AG129 mice[135]Cytidine analogAzvudine/FNCThe actual target has been identified by quantitative real-time reverse transcription-PCR (RT-qPCR), in vitro 3D^pol^ activity, and isothermal titration calorimetry (ITC) experimentsFurther studies in animal models and clinical researchRD, 293 T and MDCK cellsICR mice[136]Nucleotide triphosphate metaboliteGS-646,939The actual target has been identified by enzyme kinetics, competitive incorporation assays, structural modeling and functional validationBroad-spectrum antiviral activity against respiratory RNA viruses--[137]Pyrazine-carboxamide ribonucleotideT-1106 triphosphateThe target has been predicted by molecular dynamics simulationsTo optimize nucleoside analogs for broad-spectrum efficacy and reduce resistance and toxicityRD cellsHSCARB2 mice[173]Nucleoside phosphonate analoguesβ-D-Xylofuranosyl adenine nucleoside phosphonateThe actual target has been identified by molecular dynamics (MD) simulationsTo overcome steric hindrance during synthesis and refine interactions with RdRp active sitesCancer cells-[138]3 C and 3DNatural productPetroleum ether extract of Tournefortia sibirica L. (PE-TS)The target has been predicted by compound-target network analysis and molecular dockingTo separate effective compounds and uncover theispecific mechanismsRD cellsBALB/c mice[174]IRESN-phenylbenzamide derivativeIMB-ZThe actual target has been identified by inhibition test in vitroFurther studies in animal models and clinical researchVero and HeLa cells-[175]Antiprotozoal drugEmetineThe actual target has been identified by inhibition test in vitroTo reduce compound toxicityRD and Vero cellsKunming mice[145]Small moleculeDMA-135The actual target has been identified by pull-down experiments in cell cultureTo optimize the binding affinity and evaluate the utility as a broad-spectrum inhibitorVero and SF268 cells-[146]Natural productKaempferolThe target has been predicted by siRNA knockdown assayIdentification of molecular mechanismsRD cells-[176]TDPThe actual target has been identified by comparative proteomic profiling analysisFurther studies in animal models and clinical researchRD cells-[147]PruninThe actual target has been identified by viral resistance mutationsMore 3D RNA docking studies to affirm the mechanism and the combination of ribavirin and pruninRD cellsBALB/c mice[149]Licochalcone A (LCA)The actual target has been identified by luciferase quantitative experimentsIdentification of molecular mechanismsRD and Vero cellsICR mice[177]
Host-directed antivirals
Inhibitors of entry
The early stages of the virus lifecycle are ideal targets for preventing viral infection because, by this point, the virus has not yet multiplied. Additionally, drugs that target the host can be used as broad spectrum anti-enterovirus inhibitors [178, 179]. Cell surface receptor proteins like hSCARB-2 and HS and host factors associated with replication are promising targets for EV71 treatment. Receptor hSCARB-2 interacts with VP1 and/or VP2 capsid proteins and recognizes all EV71 strains [180]. Acarbose was hypothesized to decrease the intestinal infection of EV71 by binding to this receptor or by inhibiting various glycolic receptors on the cell surface [181].
Cell surface heat shock protein 90 (HSP90) potentially facilitates viral entry, protecting the virus from degradation by the proteasome [180]. Inhibition of HSP90 using geldanamycin (GA) and its analogue 17-allyamino-17-demethoxygeldanamycin (17AAG), or the small molecule VER-50589, effectively countered EV71 both in vitro and in vivo [182].
Nucleolin is a nucleolar phosphoprotein located in fibrillar regions of the nucleolus and on the cell surface [183]. A synthetic peptide from VP1, SP40, could bind to the RNA binding domains (RBDs) of nucleolin, and combination of anti-hSCARB-2 and SP40 led to almost complete viral inhibition (99.5%) [184].
Sertraline, an FDA-approved drug with a long clinical history of use with approval for major depression and obsessive compulsive disorder [185], may be repurposed as a EV71 antiviral. Sertraline is lipophilic and after reaching acidic compartments such as late endosomes and lysosomes, its basic amine groups undergo protonation, thus the drug is trapped in the lysosome and accumulates in acidified compartments, thus sertraline neutralizes the pH level of the endolysosomal route, crucial for entry [186]. The EV71 receptor is localized in the endolysosomal compartments and shuttles to the plasma membrane, therefore sertraline might also target the SCARB2 in the acidic milieu along the endolysosomal pathway. Whether lysosomal-resident proteins, such as ASM and SCARB2, are targets of sertraline merit further investigation. Thus, alkalinization of acidic compartments in host cells is an effective strategy for reducing viral infection and that the lysosome is a viable target organelle for antiviral drug discovery [186]. The lack of inhibitors targeting endocytosis and uncoating steps may be related to the differences in the endocytosis and uncoating steps mediated by different receptors.
Inhibitors of post-entry stage
Host factors involved in replication complex formation are also potential anti-EV71 targets. For example, phosphatidylinositol-4-kinase IIIβ (PI4KIIIβ) is a key enzyme in the phosphoinositide signalling pathway that is crucial for the replication and survival of various viruses [50]. Inhibitors of this enzyme, N-(4-methyl-5-arylthiazol)−2-amide derivatives, were identified against rhinoviruses with activity also against EV71 [187]. Compound N373 has been reported to inhibit PI4KB, improving the survival and pathology of mice when combined with viral capsid inhibitor G197 [54]. Oxysterol-binding protein (OSBP) binds PI4P inducing the exchange of PI4P in the replicating organelle membrane with cholesterol from the endoplasmic reticulum, which results in increased cholesterol content in the replicating organelle membrane [188, 189]. Also, YM201636 inhibits FYVE finger containing (PIKFYVE) kinase, blocking the endosomal sorting complex required for transport (ESCRT) and disrupting EV71 replication [190]. The small-molecule MPA-CF3, a 10,10’-bis(trifluoromethyl) marinopyrrole A derivative, exerts potent anti-EV71 activity by targeting the host factor coatomer subunit zeta-1 (COPZ1) [191]. Upon binding, this drug disrupts the interaction between COPZ1 and 2 C protein, blocking COPZ1-mediated retrograde transport of 2 C to the ER. This impairs replication organelle formation and viral RNA synthesis. As a host-directed antiviral, MPA-CF3 offers broad-spectrum potential and a high barrier to resistance, but off-target effects and cytotoxicity are a concern [191]. Future studies should focus on optimizing selectivity, validating efficacy in vivo, and exploring combination therapies with direct-acting antivirals.
Other host factors are involved in genome replication. For example, human dihydroorotate dehydrogenase (HsDHODH), a conserved mitochondrial enzyme, supplies pyrimidine nucleotides for RNA synthesis. The small molecule RYL-634 exhibited a strong binding affinity for HsDHODH and shows excellent potency against EV71 (EC_50_ = 4 nM) [192]. Another DHODH inhibitor ML390 inhibited EV71 replication by blocking de novo pyrimidine synthesis, alleviating viral replication and pathogenesis in a mouse infection model [193]. Other natural compounds targeting the post-entry stage have also been identified. Luteolin, a naturally occurring flavonoid, exhibited potent anti-EV71 and coxsackievirus A16 (CA16) activity, as shown by a high-throughput screening assay [194]. Although the target is unknown, time-of-addition studies demonstrated action at the post-attachment stage and inhibition of viral RNA replication. The peptide S78, derived from the host heat shock protein Hsp27, targets the phosphorylation of Hsp27 at Ser78, promoted during infection [195]. This phosphorylation triggers Hsp27 nuclear translocation, inducing the cytosolic redistribution of hnRNP A1. These processes subsequently inhibit viral IRES-mediated translation and replication. Peptide S78 was highly selective and had minimal cytotoxicity, but typical of peptide-based therapeutics, lacked stability and presented problems with intracellular delivery [82]. This approach highlights the potential of modulating host-virus interactions for broad-spectrum antiviral therapies.
Lipid metabolism in the post-entry stage may provide some targets for the design of antiviral drugs [196]. EV71 reportedly exploits host cell lipid β-oxidation to promote its replication, which makes lipid anabolic enzymes potential targets [197]. Drugs like C75 and TOFA inhibit two key enzymes in the fatty acid synthesis pathway, fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC), respectively, significantly inhibiting EV71 replication, whereas the carnitine palmitoyl transferase 1 (CPT1) inhibitor Etomoxir reduced triglyceride levels (lipid droplets) and inhibited EV71 replication [197]. Recently, FT895 was found to inhibit histone deacetylase 11 (HDAC11), reducing EV71 infection both in vitro and in vivo [198]. HDAC11 has been suggested to support EV71 replication by modulation of lipid droplets and increasing lipid metabolism [198]. In addition, energy generated during lipid metabolism has also been speculated to promote EV71 replication, but the specific mechanism needs to be further studied.
Interference with autotropism and apoptosis, which can be induced by EV71 infection [199], is also an effective way to block virus replication. For example, the PI3K/Akt/mTOR signaling pathway is related to EV71-induced autophagy in mice, where mTOR plays a crucial role facilitating EV71 infection post-entry [77]. Inhibition of mTOR in host cells by a novel Torin2 derivative may be an effective EV71 antiviral strategy [200]. 6-Thioguanine (6-TG), an FDA-approved anticancer agent, inhibits autophagy by targeting host BIRC3, thus suppressing EV71 replication [201]. This drug was highly selective and showed therapeutic potential for EV71-associated diseases.
During EV71-induced apoptosis, ROS generation is observed [202], therefore regulation of the cellular redox environment should protect host cells. Regulation of the Keap1-Nrf2 axis by the natural compound berberine reduced ROS production [203]. ROS generation was also reduced by ebselen, an organoselenium molecule with glutathione oxidase-like activity, thereby decreasing caspase-3 activity and apoptosis [204]. Additionally, a recent study demonstrated that the natural compound magnolol exerts anti-EV71 effects by targeting the Nrf2-SLC7A11-GSH pathway [205]. Magnolol was found to activate Nrf2, leading to increased expression of SLC7A11 and subsequent elevation of intracellular glutathione (GSH) levels. The drug suppressed ROS generation, thereby mitigating EV71-induced oxidative stress and apoptosis [205]. Saucerneol from Saururus chinensis, a medicinal herbaceous plant, has anti EV71 activity in Vero cells possibly via regulation of mitochondrial ROS (mROS) generation [206] Table 3.
Table 3. Host-directed antiviralsTarget/MechanismClassification of antiviralsName ofantiviralsStudy stagesDirection of optimizationStudy Stages/Experimental modelReferenceIn vitroIn vivoHost cellular receptorsPeptideSP81 peptideThe actual target has not yet been identifiedChemical modifications and encapsulation of peptide-based drugs in nanocarriersRD cells-[92]Glucosechannel inhibitorAcarboseThe actual target has been identified by X-ray crystallographyIdentification of molecular mechanismsRD, CCD18-Co, DLD-1, FHC and Neuro-2a cellsICR mice[181]Geldanamycin analogueVER-50,589The actual target has been identified by X-ray crystallographyStructural optimization and reduction of side effectsRD and Vero cellsICR mice[182]Synthetic peptideSP40The binding site has been identified by global dockingCo-crystallization of peptides with the receptorsRD cells-[184, 207]Cell surface glycosaminoglycans (GAGs)Polynuclear platinum complex (PPC)TriplatinNCThe actual target has been identified by inhibition test in vitroFurther validation in vivo experimentsRD cells-[208]Inhibit the interaction between viruses and heparan sulfateGlycosaminoglycan derivativeFucosylated chondroitin sulfate (FCS)The actual target has not yet been identifiedIdentification of molecular mechanismsVero cells-[209]PolyaminesSynthetic macrocyclic moleculeCucurbit[7]uril (CB[7])The actual target has not yet been identifiedIdentification of the target and molecular mechanismsRD cells-[210]Host factors responsible for EV71 RNA replicationAndrographolide DerivativesZAF-47The actual target has not yet been identifiedIdentification of the target and molecular mechanismsRD and Vero cells-[211]AKT1, ALB and SRCTraditional Chinese herbal formulaChangyanning (CYN) TabletsThe actual target has been identified by molecular dockingFurther validation in vivo experimentsRD cells-[212]Compete with naturally occurring nucleotides for incorporation into viral RNAFluorinated purine analogueFludarabine (2 F-ara-A)The actual target has not yet been identifiedFurther validation in vivo experiments and reduction of toxicityVero, BHK21, U251 MG and HMC3 cells-[213]LysosomesNatural productEnanderinanin JThe actual target has not yet been identifiedIdentification of the target and molecular mechanismsVero cells-[214]Pim1Synthesized compoundsCX-6258The actual target has been identified by structure-activity relationship analysisTo determine whether Pim1 directly binds eIF4G or affects eIF4G phosphorylationRD, HeLa and 293T cells-[215]Alpha-enolase (ENO1)Glycolysis inhibitorDichloroacetic acid (DCA)The actual target has not yet been identifiedIdentification of the target and molecular mechanismsRD cells-[216]Host methyltransferase METTL3Nucleoside analoguesCompound 2, 11b and 17The actual target has been identified by molecular dockingIdentification of reasons for the marked selectivity of these agentsRD cells-[217]Oxidative stress-mediated ERS/autophagy pathwayNatural product with nanoparticlesResveratrol-loaded nanoparticles (RES-NPs)The actual target has not yet been identifiedIdentification of the target and molecular mechanismsRD cells-[218]Rho Associated Coiled-Coil Containing Protein Kinase 1(Rock 1)Kinase inhibitorGSK269962AThe actual target has been identified by expression of purified proteins in vitroIdentification of molecular mechanisms and evaluation of safetyRD cells-[219]Ameliorate the decrease of caspase-3 and PARPNatural productTotal astragalosides (ASTs)The actual target has not yet been identifiedIdentification of the target and molecular mechanismsGES-1 cells-[220]Raf-MEK-ERK signaling pathwayClinically approved B-Raf inhibitorVemurafenibThe actual target has been identified by structure-guided chemistry platformIdentification of molecular mechanismsRD cells-[221]PI3K/AKT/mTOR and NF-κB signaling pathwaysTraditional Chinese herbal formulaPien Tze Huang (PZH)The actual target has not yet been identifiedIdentification of the target and molecular mechanismsRD and Vero cellsICR mice[222]AKT Signaling PathwayNatural productSalvianolic Acid BThe actual target has not yet been identifiedIdentification of the target and molecular mechanismsHeLa cells-[223]P53 and STAT1 signaling pathwayPolysaccharidDurvillaea Antarctica Polysaccharide (DAPP)The actual target has not yet been identifiedIdentification of the target and molecular mechanismsVero cells-[224]Inhibit acid sphingomyelinase (ASM)AntidepressantSertralineThe actual target has not yet been identifiedIdentification of the target, the molecular mechanisms and antiviral spectrumRD and HeLa cells-[186]PI4KB inhibitorSynthesized compoundCompound N373The actual target has been identified by biochemical assayFurther validation in vivo experimentsRD cellsBALB/c mice[54]Disrupt the EV71 replication complexInhibitor of PIKFYVE kinaseYM201636The actual target has been identified by inhibition test in vitroIdentification of molecular mechanismsRD cells-[190]DHODH inhibitorSmall moleculeRYL-634The actual target has been identified by chemical rescue experimentLead optimization to give more drug-like analoguesRD cells-[192]Small moleculeML390The actual target has been identified by genetic resistance and sequencing effortsChemical modifications to reduce plasma protein-binding activity and toxicityRD, HeLa and Vero cellsICR mice[193]Ser78 phosphorylation site of Hsp27PeptidePeptide S78The actual target has been identified by in vitro and cell-based assaysTo Enhance peptide stability, membrane permeability and specificity; reduce off-target effects and toxicityHEK293 T and RD cells-[195]Inhibit key enzymes in the fatty acid synthesis pathwayChemical reagentC75 and TOFAThe actual target has been identified by inhibition test in vitroFurther validation of the specific link in the viral replication cycle that energy is utilizedRD and HUVEC cells-[197]CPT1 inhibitorChemical reagentEtomoxirThe actual target has been identified by inhibition test in vitroFurther validation of the specific link in the viral replication cycle that energy is utilizedRD and HUVEC cells-[197]HDAC11 inhibitorAnalog of N-hydroxy-tetrahydroisoquinoline-7-carboxamideFT895The actual target has been identified by bioluminescence resonance energy transfer (BRET) target engagement assayIdentification of molecular mechanismsHT-29, HeLa and Vero cellsC57BL/6 mice[198]PI3K/Akt/mTOR signaling pathwayTorin2 derivativeCompound 11eThe actual target has been identified by molecular modeling studyBetter water solubility and potent activityRD cells-[200]Keap1-Nrf2 axisNatural productBerberine (BBR)The actual target has been identified by inhibition test in vitroIdentification of molecular mechanismsU251, SK-N-MC and A549 cellsICR mice[203]Reduce ROS generationOrganoselenium moleculeEbselenThe actual target has been identified by inhibition test in vitroIdentification of the mechanism and the connection between EV71-mediated oxidative damage and ebselen’s antioxidant activityVero cells-[204]Nrf2-SLC7A11-GSH pathwayNatural productMagnololThe actual target has not yet been identifiedThe extent to which magnolol modulates levels of innate immune-related genes and proinflammatory factors at the cellular level.RD and HeLa cellsICR mice[205]Regulate mitochondrial ROS (mROS) generationNatural productSaururus chinensisThe actual target has been identified by inhibition test in vitroClinical trials and assessment of efficacy and safetyVero cellsBALB/c mice[206]PI3K-AKTNatural productAstragaloside IV (AST-IV)The target has been predicted by network pharmacologyIdentification of the targeted binding of AST-IV to PI3K and AKT and further validation in vivo experimentsNormal human gastric epithelial (GES-1) and RD cells-[225]Coatomer subunit zeta-1 (COPZ1)10,10′-bis(trifluoromethyl) marinopyrrole A derivativeMPA-CF3The actual target has been identified by quantitative chemo-proteomicsVivo efficacy evaluationRD and Vero cells-[191]Inhibit EV71 replication at the early stages of the viral cycleScorpion venom antimicrobial peptide derivativeBmKn2-T5The actual target has not yet been identifiedFurther understanding of its structural and functional properties, and the design of pharmacologically active peptides with high activity and selectivityRD and Vero cells-[226]Reduce the expression of baculoviral IAP repeat containing 3 (BIRC3) and attenuate BIRC3-mediated the complete autophagyA thiopurine drug6-thioguanine (6-TG)The actual target has been identified by RNA-sequencing analysis combined with siRNA knockdown, overexpression experiments, and functional validationTo enhance anti-EV71 activity, improve selectivity and reduce cytotoxicityHT-29 (human colorectal cell lines), HeLa and Vero cells-[201]
Conclusions and perspectives
Targeting host factors has the advantage that emergence of resistance is unlikely and common mechanisms ensure wide applicability to different viruses. However, targeting host factors is inherently dangerous because these targets must remain minimally functional. In contrast, viral targets are in principle more specific to certain strains and have the problem of resistance, especially in RNA viruses. Thus, it is clear that only a combination of treatments can succeed, and only if this treatment is sufficiently short to prevent emergence of resistance. Such combinations, which leverage both viral- and host-targeting agents, can synergistically enhance efficacy while reducing individual drug doses to mitigate toxicity. Effective drugs are available to combat EV71. Possibly the most promising are those that target the capsid, especially when the virus still has not entered the cell. However, since treatment is administered usually after infection, these drugs should be able to penetrate the cell membrane, which is more difficult for peptides. Additionally, peptides are easily hydrolyzed, therefore cyclic peptides, which are resistant to thermal and enzymatic degradation, should be explored. Emerging computational tools can further accelerate the design of such peptides by predicting their stability and binding affinity to viral targets [227].
Higher affinity and lower toxicity can only be optimized when high resolution structures are available by means of X-ray diffraction, solution or solid-state NMR, or cryo EM methods. Even when the target and binding site is known, in the absence of this structural data optimization cannot be performed, and the value of purely in silico methods is questionable.
Beyond the enzymatic activity of some of the proteins in EV71, more attention should be paid to proteins for which we do not have structural data. These proteins have been reported to have channel activity and membrane remodeling activity, which probably depends on oligomerization. These are activities that can be targeted by small molecules, or more likely peptides.
Other alternative compounds such as flavonoids show anti EV71 activity [228] and nanodrug delivery and chemical modifications could be used to enhance bioavailability and potency [170, 229], but detailed structural data is missing.
Translating these candidates to clinical use faces challenges, including the need for age-appropriate formulations, scalable production, and rigorous safety assessment in pediatric populations. In these populations, immature organ function and rapid growth demand tailored dosing strategies.
Notably, given EV71’s propensity to invade the central nervous system, strategies to enhance drug delivery across the blood-brain barrier—such as ligand-functionalized nanocarriers or targeted delivery systems—warrant further exploration to mitigate severe neurological complications. Integrating these advances with a focus on pediatric safety and clinical translatability will be pivotal to advancing EV71 therapeutics.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Liu Y, Lv P, Wang W, Zhang J, Zhou X, Qiu Y et al. Structural insight into EV-A 71 3A protein and its interaction with a peptide inhibitor. Virol Sin. 2023;S 1995-820X(23)00112-8.10.1016/j.virs.2023.09.004PMC 1078665737757951 · doi ↗ · pubmed ↗
- 2Mj HT, Mg N, Cw M, Jj A, Fw D et al. S,. A second virus-encoded proteinase involved in proteolytic processing of poliovirus polyprotein. Cell [Internet]. 1986 [cited 2024 Jun 2];45. Available from: https://pubmed.ncbi.nlm.nih.gov/3011278/10.1016/0092-8674(86)90790-73011278 · doi ↗ · pubmed ↗
- 3Yuan J, Shen L, Wu J, Zou X, Gu J, Chen J et al. Enterovirus A 71 Proteins: Structure and Function. Front Microbiol [Internet]. 2018 [cited 2025 Feb 16];9. Available from: https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2018.00286/full 10.3389/fmicb.2018.00286 PMC 582639229515559 · doi ↗ · pubmed ↗
- 4M A, T W, H S. Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. The Journal of general virology [Internet]. 2008 [cited 2024 Jun 2];89. Available from: https://pubmed.ncbi.nlm.nih.gov/18796721/10.1099/vir.0.2008/002915-018796721 · doi ↗ · pubmed ↗
- 5Olasunkanmi OI, Fei Y, Avala Ntsigouaye J, Yi M, Wang Y, Liu J et al. Antiviral activity of trans-Hexenoic acid against coxsackievirus B and enterovirus A 71. Antimicrob Agents Chemother. 2023;67(3):e 00868–22.10.1128/aac.00868-22PMC 1001928936786598 · doi ↗ · pubmed ↗
- 6Zhang Q-Y, Li J-Q, Li Q, Zhang Y, Zhang Z-R, Li X-D, et al. Identification of Fangchinoline as a broad-spectrum enterovirus inhibitor through reporter virus based high-content screening. Virol Sin. 2024;39(2):301–8.10.1016/j.virs.2024.02.006PMC 1107463738452856 · doi ↗ · pubmed ↗
- 7Cipriani A, La Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, et al. Sertraline versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2010;1:CD 006117.10.1002/14651858.CD 00611720091586 · doi ↗ · pubmed ↗