The first complete mitochondrial genome sequences of an ancient orphan evolutionary lineage of Eumolpinae leaf beetles endemic to the South Pacific
Anabela Cardoso, Jesús Gómez-Zurita

TL;DR
This study reports the first complete mitochondrial genomes of three leaf beetle species from an ancient lineage in the South Pacific, enhancing our understanding of insect evolution.
Contribution
The study provides the first complete mitogenomes from an orphan evolutionary lineage of Eumolpinae leaf beetles, enriching phylogenetic data for this poorly represented group.
Findings
The mitogenomes retain ancestral gene order with a synapomorphic switch in trnA and trnR positions.
Nucleotide composition and codon usage patterns suggest mutation pressure rather than selection as the main driver.
Phylogenetic analysis supports prior classifications and highlights rapid diversification in Cryptocephalinae.
Abstract
There are very limited mitogenome data representing most animal groups, particularly among the insects, which are otherwise extremely diverse and fulfill important ecological, sanitary, forest and agroeconomic roles. Increasing taxonomic diversity with new additions to the pool of mitogenomes for unrepresented evolutionary lineages is an opportunity to increase the phylogenetic power of mitogenome data, as well as refining our understanding of mitogenome diversity. Here, we characterize the complete mitogenomes of three species in two subgenera of Taophila leaf beetles, members of the hyperdiverse Eumolpinae, currently very poorly represented by mitogenomes, and the first representing an enigmatic independent evolutionary branch of the Eumolpini tribe, originated in the Cretaceous-Paleogene transition and endemic to the South Pacific. These mitogenomes, assembled from genomic Illumina…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
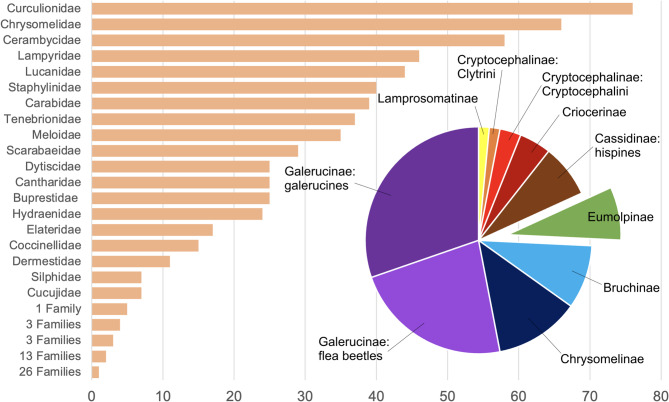 Figure 1
Figure 1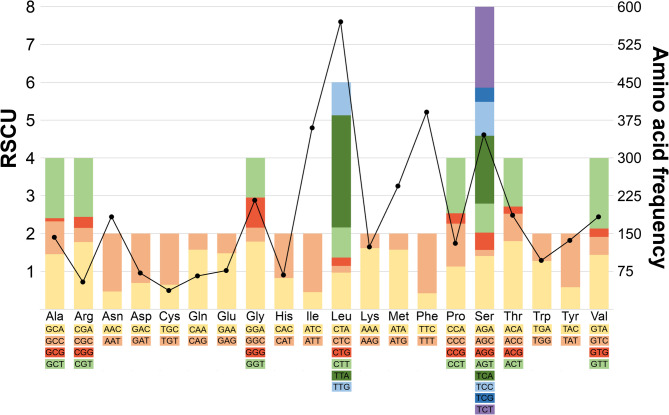 Figure 2
Figure 2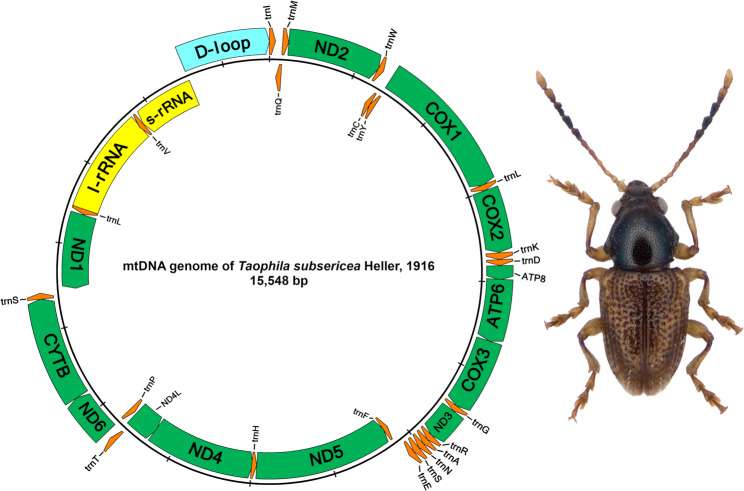 Figure 3
Figure 3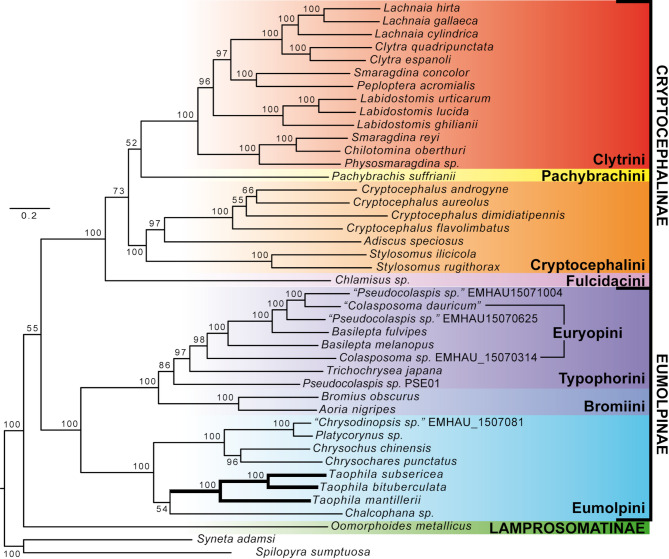 Figure 4
Figure 4- —https://doi.org/10.13039/501100011033Agencia Estatal de Investigación
- —Unidad de Recursos de Información Científica (URICI-CSIC)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsColeoptera Taxonomy and Distribution · Genomics and Phylogenetic Studies · Coleoptera: Cerambycidae studies
Introduction
The use of mitochondrial DNA (mtDNA) sequences for genetic and evolutionary studies in animals has a very long tradition [1]. But in the era of genomics, the ease of assembling complete or nearly complete mitogenomes as either byproducts or end-targets of genomic or metagenomic DNA sequencing using Next Generation Sequencing technologies has propelled the accumulation of mitogenome data for a large variety of organisms across the Metazoan tree of life (e.g., [2–4]). Currently, public sequence databases such as GenBank (28 November, 2024; https://www.ncbi.nlm.nih.gov/datasets/organelle/) host complete mitogenomes of 16,057 nominal species of animals, although they are not representative of Metazoan diversity, with nearly 49.0% of these data from chordates, and only 31.1% from insects, while these two animal classes represent approximately 3–6% and 75% of known species, respectively [5]. Moreover, there are also strong biases in species coverage within animal groups. For example, based on these same data, in the case of underrepresented insects, some 21.6% of available reference mitogenome data actually correspond to different species of Lepidoptera, while a mere 14.0% and 5.1% represent the inordinately diverse beetles and hymenopterans, with some 702 and 256 species with characterized mitogenomes (between 1.5–2.0‰ of described species), respectively. The precision of interpretations based on GenBank data mining may be affected by the vagaries of mining strategies but also annotation and taxonomic inaccuracy, but support to this bias comes from an independent, detailed review of insect mitochondrial genomics by S. L. Cameron [4], reaching similar estimates and conclusions based on data collected one year prior to ours. Data accumulates rapidly, but there are still huge gaps in our representation of the animal tree of life when it comes to mitogenomes. In this respect, mitophylogenomics, phylogenetic studies based on the full or most information available in mitogenomes, has proven a useful approach to increase the diversity of available mitogenomes, because of their interest in representing taxonomic diversity within and around the focal taxonomic group (e.g., [6–10]). At the same time, the utility of mitophylogenomics is compromised by uneven taxon coverage and, as these data become comparable in terms of taxonomic representation to standard phylogenetic markers for most groups, the more power and support will have the results based on these approaches. Hybrid approaches that combine complete or partial mitogenomes and nuclear genes have provided increased phylogenetic support in some cases (e.g., [11, 12]). Acknowledging these biases and limitations highlights the importance of progressively filling the taxonomic gaps also for this type of data.
When preparing this study, beetle mitogenomes tagged as reference genomes in GenBank—admittedly an underestimation, but taken as representative of existing data—are available for 65 families of Coleoptera, with 89% of these data belonging to 19 families only (Fig. 1). The bulk of these data were sequenced in dedicated mitophylogenomic studies of particular groups, such as fireflies (Lampyridae; [13]) or Onthophagini scarabs [14], or studies focusing on beetle species generally associated with stored products, agriculture or timber damage, including skin beetles (Dermestidae). The three largest phytophagous beetle families dominate available mitogenomes: Curculionidae, Chrysomelidae and Cerambycidae. With 65 reference mitogenomes (representing less than 1.6‰ of species in this family; [15]), leaf beetles are the second most sequenced beetle group, with many species studied due to their pest status, and a significant proportion have been characterized in the framework of mitophylogenomic studies (e.g., [16, 17]). Nie et al. [16]. focused on the largest subfamily of leaf beetles, the Galerucinae, representing more than half of available chrysomelid mitogenomes (Fig. 1), while other subfamilies have a low representation. Galerucinae and their sister group Chrysomelinae [18] include serious crop pests and this may explain, in part, why these subfamilies are the most common target of mitogenomic sequencing. For example, out of 52 leaf beetle species represented in the reference genome database in GenBank (3 December, 2024; https://www.ncbi.nlm.nih.gov/datasets/genome/), up to 44 were from species in these two subfamilies.
Fig. 1. Reference mitogenomes of Coleoptera. Number of reference mitogenomes available for different families of Coleoptera in GenBank (November, 2024), and proportions of mitogenomes corresponding to different subfamilies (or suprageneric assemblages) of the Chrysomelidae. Eumolpinae, with five reference mitogenomes, and their closest relatives Cryptocephalinae and Lamprosomatinae, are poorly represented in public genome reference databases
Eumolpinae stand out among the genomically less studied groups of leaf beetles, possibly because of their enormous diversity, with an estimated 7,000 described species [15], their mostly tropical distribution, and a general lack of taxonomic resources for the group. To date, there are complete mitochondrial genome sequences of Eumolpinae for five Asian species considered pests (Fig. 1), including Aoria nigripes (Baly, 1860) [19], Basilepta fulvipes (Motschulsky, 1860) [20] and B. melanopus Lefèvre, 1893 [21], Colasposoma dauricum auripenne (Motschulsky, 1860) [22], and Trichochrysea japana (Motschulsky, 1858) (Huang & Wu, unpublished data). Public nucleotide sequence databases, apart from a few unpublished sequences, also host nearly complete, partially annotated sequences for Chalcophana sp. [17], Chrysochares punctatus (Gebler, 1845) [23], Chrysochus chinensis Baly, 1859 [17], Chrysodinopsis sp. [24], Platycorynus sp. [17], and Pseudocolaspis sp. [25]. Several molecular phylogenetic studies have found a close association between Eumolpinae and the subfamilies Lamprosomatinae and Cryptocephalinae [17, 18, 26, 27], which are also poorly represented for genomic and mitogenomic resources (Fig. 1). A clade of these three subfamilies is also supported by a derived mitogenome arrangement [17], a synapomorphy consisting of the trnR and trnA genes having swapped positions relative to the arrangement found in the ancestral insect and retained by all other Chrysomelidae subfamilies sequenced to date.
The genus Taophila Heller, 1916 is an endemic genus of Eumolpinae from New Caledonia. This genus belongs to the tribe Eumolpini and is member of an enigmatic orphan lineage of this tribe endemic to and highly diversified in the South Pacific, from Vanuatu and New Caledonia to the South Island of New Zealand, also present in Fiji and Kermadec islands. The evolutionary enigma behind this South Pacific clade is the lack of clear shared ancestry with other groups, diverging from the rest of Eumolpini some 43.0–75.2 Ma ago [28]. One possible explanation for this orphan lineage is their origin in an evolutionary branch established in Antarctica but gone extinct at the end of the Eocene; however, sampling artifacts were not to be discarded [28]. Taophila has 21 species, one in the subgenus Jolivetiana Gómez-Zurita & Cardoso, 2014, and the rest in the nominotypical subgenus [29]. The genus has some rather interesting biological attributes for evolutionary and ecological studies, including striking sexual dimorphism, with females displaying the most distinctive morphological traits among species [29]. Taophila also includes one of the few documented cases of trophic associations with ferns in the leaf beetles [30, 31]. As mentioned, previous molecular systematics attempts at finding the evolutionary ties of South Pacific Eumolpini with other relatives worldwide were unsuccessful [28]. Considering that taxonomic density for mitophylogenomic studies will increase more rapidly than nuclear or genomic information [4], the availability of high-quality mitogenomic data for this lineage shall increase the chances of eventually finding their closest relatives, the missing link, or support the hypothesis of extinct relatives, possibly in Antarctica. To this end, in this work, we contribute complete, fully-annotated mitochondrial genomes for three species in both subgenera of Taophila. These data add one evolutionary branch previously missing from the mitophylogenomic tree of the Eumolpinae. Nucleotide composition and codon usage features of these new mitogenomes are described and the new mitogenomic data used to infer the phylogenetic history of the Eumolpinae, Cryptocephalinae and Lamprosomatinae.
Materials and methods
Samples and sequencing
Non-destructive genomic DNA extractions obtained with the DNeasy Blood & Tissue kit (Qiagen Iberia, Madrid) from a previous study [31], were available for three species of Taophila, including the type species, T. subsericea Heller, 1916 (specimen JGZC-NC103), T. bituberculata Platania & Gómez-Zurita, 2022 (specimen JGZC-NC102) and T. mantillerii Jolivet, Verma & Mille, 2007 (specimen JGZC-NC091), the type of the monotypic subgenus Jolivetiana Gómez-Zurita & Cardoso. Library preparation and sequencing were externalized to the Sequencing Unit at CNAG (National Center for Genomic Analyses, Barcelona, Spain). Extracted DNA was quantified using Qubit (Thermo Fisher Scientific Inc., Waltham MA) and Femto Pulse (Agilent Technologies Spain, Madrid) technologies, yielding total DNA amounts between 0.18 and 0.52 µg. Genomic DNA was sheared on a LE220-Plus Focused ultrasonicator (Covaris Ltd., Brighton, UK) and short-insert paired-end libraries were obtained with the PCR-free protocol of KAPA HyperPrep Kit (Roche Sequencing Solutions, Inc., Pleasanton CA), including end-repair, adenylation and ligation to Illumina-compatible adaptors with unique dual indexes and unique molecular identifiers (Integrated DNA Technologies, Leuven, Belgium). Sequencing was performed in paired-end mode (2 × 151 bp) on two independent flow cells on a NovaSeq 6000 Sequencing System (Illumina, San Diego CA), and raw data processing, including image analysis and FASTQ generation, were done with the manufacturer’s Real-Time Analysis software, RTA version 3.4.4. Data obtained from each flow cell were analyzed independently to assess the performance of the assembly and annotation procedure. Voucher specimens and DNA aliquots are kept in the authors collections (IBB, CSIC, Spain) and raw sequence data are available under the European Commission Open Data Repository, Zenodo (10.5281/zenodo.16900841).
Mitogenome assembly and annotation
Data quality control and filtering, assembly and annotation were performed with MitoZ version 3 [32]. MitoZ is a Python pipeline that completes raw paired-end reads filtering and cleaning with a custom Perl script; read assembly with MEGAHIT [33]; filtering of false mitochondrial scaffolds and NUMTs based on HMMER, a profile Hidden Markov Model approach [34] and Tiara classification of sequences [35]; and annotation using tBLASTn and Genewise [36] for protein-coding genes (PCG), MiTFi [37] for tRNAs, and Infernal [38] for rRNAs. MitoZ assembly and annotation used software defaults, except that a maximum of 4 or 5 Gb for raw data (equivalent to 26.49 M or 33.11 M reads) were used in assemblies, “Arthropoda” was specified as taxonomic source to guide the selection of correct mitochondrial genome scaffolds and “Coleoptera” for annotation steps. A multi-Kmer approach for genome assembly was applied with K = 79, 99 and 119. Complete, circularized mitogenomes inferred by MitoZ have random initiation points, and sequences were reoriented to a standard gene order manually in Geneious Prime version 2024.0.7 (Dotmatics, Boston MA). The complete annotated mitogenomes of Taophila were deposited in GenBank under accession numbers PV240303 (T. bituberculata), PV240304 (T. mantillerii) and PV240305 (T. subsericea).
Analysis of protein-coding gene features
Structural features of annotated mitogenomes, including gene length, nucleotide composition, and start/stop codons, were extracted in Geneious Prime. Alternative start and stop codons and incomplete stop codons lack empirical verification, and were deduced computationally and based on comparisons with published mitogenome annotations of close relatives. Codon usage bias was estimated via relative synonymous codon usage (RSCU) metrics, from a concatenation of the protein-coding genes using CAIcal [39], and the effective number of codons metric (Nc; [40]) with CodonW version 1.4.4 (written by John Peden, and implemented in the Galaxy server; [41]). Nucleotide composition bias was estimated via correlation of G + C content at combined first and second codon positions (GC12) versus third codon positions (GC3); this correlation can distinguish the role of mutation pressure versus natural selection in nucleotide composition [42]. The fit of these two variables to normal distributions was assessed based on the Shapiro-Wilk normality test and homoscedasticity on the Breusch-Pagan test. The significance of correlation was estimated using the Pearson correlation test. All tests were computed with the ggplot2, ggpubr and car packages in R version 4.3.1 [43].
Mitophylogenomics
Complete or nearly complete annotated mitogenomes of Cryptocephalinae, Lamprosomatinae and Eumolpinae were obtained from GenBank, together with the mitogenomes of representatives of two subfamilies traditionally associated with Eumolpinae, and considered sister to the clade composed of the three subfamilies of interest [18, 27, 44], selected here as outgroups: Spilopyra sumptuosa Baly, 1860 (Spilopyrinae; [17]) and Syneta adamsi Baly, 1877 (Synetinae; [17]). Sequences availability is listed in Table 1. The 13 protein-coding and two rRNA genes of these leaf beetle mitogenomes were aligned independently using MAFFT 7 algorithms, L-INS-i for protein-coding genes and Q-INS-i for rRNA genes [47]. Ragged ends in some alignments (nad1, nad3 and rRNA genes) were removed manually prior to the concatenation of individual alignments. The concatenated matrix was analyzed under maximum likelihood in RAxML NG [48] and Bayesian inference in MrBayes version 3.2.7 [49] with unlinked independent GTR + I + G substitution models for rRNA genes, third codon positions of protein-coding genes, and combined first and second codon positions of these genes, for a total of 28 independent data partitions. Maximum likelihood phylogenetic inference consisted of 20 individual runs starting from random topologies, with branch support (BS) assessed through bootstrap search with 500 pseudoreplicates. Bayesian inference consisted of two independent runs of one cold and three heated chains (default temperature = 0.2) for 15 M generations each, sampling parameters and trees every 1,000 generations (average standard deviation of split frequencies between runs dropped below 1% after 700 K generations). Tree topologies and node posterior probabilities (PP) were summarized excluding the initial 25% of saved trees. The optimal maximum likelihood tree and the maximum clade credibility Bayesian tree were rooted secondarily to the outgroups, Spilopyrinae and Synetinae.
Table 1. Taxon sampling for the mitophylogenomic analysis of the Eumolpinae clade and closest relativesSubfamilySpeciesAccessionReferenceCryptocephalinae FulcidaciniChlamisus sp.MK049868[17] ClytriniChilotomina oberthuri KX943432[45]Clytra espanoli KX943426[45]Clytra quadripunctata KX943461[45]Labidostomis ghilianii KX943477[45]Labidostomis lucida KX943488[45]Labidostomis urticarum MK049877[17]Lachnaia cylindrica KX943427[45]Lachnaia gallaeca KX943443[45]Lachnaia hirta KX943433[45]Peploptera acromialis HQ232809[46]Physosmaragdina sp.MK049873[17]Smaragdina concolor KX943498[45]Smaragdina reyi KX943492[45] CryptocephaliniAdiscus speciosus NC_066821UnpublishedCryptocephalus androgyne KX943411[45]Cryptocephalus aureolus KX943487[45]Cryptocephalus dimidiatipennis NC_066822UnpublishedCryptocephalus flavolimbatus MK049867[17]Stylosomus ilicicola KX943463[45]Stylosomus rugithorax KX943428[45] PachybrachiniPachybrachis suffrianii KX943489[45]Eumolpinae BromiiniAoria nigripes NC_065028[19]Bromius obscurus KX087249UnpublishedPseudocolaspis sp.KY039144[24]Pseudocolaspis sp.KY039145[24]Pseudocolaspis sp.JX412756[25] EuryopiniColasposoma dauricum KY039104[24]Colasposoma sp.KY039108[24] EumolpiniChalcophana sp.MK049868[17]Chrysochares punctatus MN745103[23]Chrysochus chinensis MK049871[17]Chrysodinopsis sp.KY039111[24]Platycorynus sp.MK049872[17]Taophila bituberculata PV240303This studyTaophila mantillerii PV240304This studyTaophila subsericea PV240305This study TypophoriniBasilepta fulvipes MT627597[20]Basilepta melanopus OP115728[21]Trichochrysea japana OR387477UnpublishedLamprosomatinaeOomorphoides metallicus MK085754[17]SpilopyrinaeSpilopyra sumptuosa MK049875[17]SynetinaeSyneta adamsi MK049876[17]
Results
Characteristics of Taophila mitogenomes
The MitoZ pipeline used approximately 19–61% of the raw read content of the original libraries, which provided nonetheless abundant high-quality sequence data for mitogenome assembly in every case, both in terms of contigs representing this genome compartment and the read depth per individual position of the assembled mitogenomes (Table 2). Duplicate mitogenome assemblies of different Illumina runs at the same individual’s DNA produced identical assembled sequences in all cases, except for differences in circularization in the control region, showing a duplication of a tract of 36 bp in one T. mantillerii assembly, of 50 bp in one T. subsericea assembly, and a tandem duplication of this same segment, adding 100 bp in one assembly of T. bituberculata (Table 2). By removing these artefactual duplications, the genomes were 15,597 bp in length in T. mantillerii, 15,498 bp in T. subsericea, and 15,484 bp in T. bituberculata. Gene order and orientation of all three Taophila mitogenomes were identical to those found in the published mitogenomes of other Eumolpinae, Cryptocephalinae and Lamprosomatinae (Fig. 2).
Table 2. Illumina library and Taophila mitogenome assembly characteristics, including data on number and quality of reads, assembled contigs and inferred genome source (based on Tiara), and sequencing depthT. bituberculataT. mantilleriiT. subsericeaLibrary 1Library 2Library 1Library 2Library 1Library 2Library reads (M)43.33138.3448.31139.8155.87125.02Total reads used (M)^a^26.4933.1126.4926.4926.4933.11Reads filtered (M)26.1832.7825.9425.9226.2032.77Q3093.6493.4493.4993.0294.2593.76Insert size peak (bp)267266151151268262Megahit Contigs21,60324,51017,78022,66125,41232,872Eukarya18,05220,41514,61018,58921,04827,201Organelle80189866190110131339Mitochondrion218237195265263367Minimum depth^b^189231335587807Maximum depth^b^320757213796353555166173Average depth^b^666.5 ± 255.23845.8 ± 395.73674.5 ± 170.53680.7 ± 156.51890.6 ± 208.541121.6 ± 221.92Length15,58415,48415,63315,59715,49815,548^a^ In two cases, MitoZ failed to circularize the mitogenomes, and the problem was solved by increasing the amount of initial data, from 4 to 5 Gb, the maximum allowed by the pipeline^b^ The extremes were always found in the control region, and depth average values were representative of the whole coding region
Fig. 2. Complete mitogenome of Taophila subsericea Heller
Characteristics of protein-coding genes
The absolute proportions of amino acids coded for in protein-coding genes of the mitogenome of Taophila subsericea, as well as the RSCU for each amino acid are shown in Fig. 3. The other Taophila mitogenomes show the same pattern with small variations (Fig. S1). Just five of 20 amino acids represented more than half (51.9%) of all the coded amino acids in the mitogenome: the polar uncharged Serine, and the hydrophobic Leucine, Phenylalanine, Isoleucine and Methionine. The five least used amino acids (8.0%) were the electrically charged Aspartic Acid, Arginine and Histidine, and the polar Glutamine and special Cysteine. The most utilized start codons in Taophila were ATN (except ATA), but also TTG, used by nad1-nad3. The most common stop codon was TAA, although TAG or truncated stop codons (T– and TA-) were identified as well (Table 3). Synonymous codon usage was always biased towards codons ending in an A or T base (Fig. 3). The G + C contents of third (W = 0.96939, p-value = 0.360) and combined first and second (W = 0.94678, p-value = 0.064) codon positions fitted normal distributions (Fig. S2), and the variances of both variables were similar (Chi-Squared = 0.21597, d.f. = 1, p-value = 0.642). The correlation between GC12 and GC3 (Fig. S2) was significant (correlation coefficient = 0.4358, t = 2.9451, d.f. = 37, p-value = 0.0056).
Fig. 3. Codon usage in the mitogenome of Taophila. Stacked column chart representing relative synonymous codon usage of the mitogenome of Taophila subsericea Heller, and line graph showing the absolute frequency of each amino acid in the set of protein-coding genes in this mitogenome
Table 3. General characteristics of protein coding genes of the mitogenomes of three species of Taophila leaf beetlesGeneLengthAA%GC^a^GC3^a^Starts. str.StartJolivetianaStops. str.StopJolivetiana atp6 67522427.4/28.6/31.120.1/20.1/23.7ATG (M)TAA atp8 1565118.6/19.2/23.111.8/17.6/15.7ATT (I/M)TAA cox1 154551435.0/37.5/34.522.8/29.4/20.4ATT (I/M)TAA cox2 ^b^ 68422729.7/31.6/32.015.0/20.7/19.2ATT (I/M)ATC (I/M)TAAT-- cox3 78526130.3/34.0/33.214.2/27.6/25.7ATG (M)TA- cytb 114037931.1/35.7/32.221.1/30.6/20.8ATG (M)TAA nad1 95131625.2/28.4/26.013.6/22.5/17.7TTG (L/M)TAG nad2 101133625.9/26.8/27.420.2/21.4/22.3TTG (L/M)TAA nad3 35411726.0/29.1/27.721.4/29.1/23.9TTG (L/M)ATC (I/M)TAA nad4 132744222.5/26.5/25.113.3/22.6/20.8ATG (M)T--TAG nad4l 2889522.6/22.6/24.018.9/18.9/17.9ATG (M)TAA nad5 171457122.6/26.0/23.511.9/20.0/13.8ATT (I/M)T-- nad6 48916221.1/24.5/21.916.7/22.8/17.3ATT (I/M)ATC (I/M)TAA^a^ Total and third codon position G + C content is given for T. bituberculata/T. subsericea/T. mantillerii^b^ In T. mantillerii, the annotation extended the cox2 gene 4 nt and 2 extra aa’s
The effective number of codons (Nc) per gene showed a strong to moderate bias, with values ranging between 30.49 and 46.51 in T. bituberculata, 32.79 and 42.19 in T. mantillerii, and 29.35 and 47.60 in T. subsericea (Table 4). In all three mitogenomes, NADH dehydrogenase subunit genes typically showed more biased values compared to the other genes.
Table 4. Effective number of codons in protein-coding genes of three mitogenomes of TaophilaGeneT. bituberculataT. mantilleriiT. subsericea atp6 36.4141.3639.38 atp8 46.51-29.35 cox1 41.9338.8343.41 cox2 38.1640.4542.98 cox3 37.3739.7047.60 cytb 37.1338.4144.36 nad1 34.9734.0538.40 nad2 37.8342.1939.61 nad3 32.0438.1937.37 nad4 30.4935.0538.92 nad4l 32.3738.0234.91 nad5 30.9536.2634.67 nad6 35.1532.7939.90
Mitophylogeny of the Eumolpinae clade
The mitophylogeny of Eumolpinae and closest relatives was consistent with previous hypotheses of relationships in this group, where the three subfamilies formed a basal polytomy (Figs. 4 and S3). The Cryptocephalinae were well supported (BS = 100%, PP = 1.00) with moderate support (BS = 73%) in the maximum likelihood tree for the Fulcidacini as sister of the remaining tribes, which formed a polytomy of monophyletic Cryptocephalini (BS = 100%), Pachybrachini (monotypic) and Clytrini (BS = 100%). In the Bayesian tree, these relationships were fully resolved as (Fulcidacini, (Pachybrachini, (Cryptocephalini, Clytrini))) (Fig. S3). All genera within Cryptocephalinae with more than one species sampled were monophyletic, with the exception of Smaragdina, also consistent with previous studies [18, 27]. Finally, the Eumolpinae clade was well supported (BS = 100%, PP = 1.00) with two strongly supported groups (BS = 100%, PP = 1.00), consistent with a clade of Bromiini, Typophorini and Euryopini, and one of Eumolpini, a topology consistent with previous phylogenies of this subfamily [18, 28, 44]. The genera Basilepta,* Colasposoma* and Pseudocolaspis in the first clade were not monophyletic. The new Taophila mitogenomes formed a monophyletic group (BS = 100%, PP = 1.00) within the Eumolpini clade, one of three well supported clades within a polytomy of Eumolpini lineages, in both the maximum likelihood and Bayesian trees.
Fig. 4. Mitophylogenomics of the Eumolpinae clade. Optimal maximum likelihood tree (L = −282,375.230644) based on protein-coding genes and rRNA data from the mitogenomes of Cryptocephalinae, Eumolpinae and Lamprosomatinae. Branch support is shown as bootstrap percentages, the lineage of new mitogenomes shown with thickened branches, and clades consistent with the subfamily and tribe systematics of leaf beetles are highlighted
Discussion
Average GenBank reference mitogenome size in insects (accessed 28 November, 2024; https://www.ncbi.nlm.nih.gov/datasets/organelle/) is 15,801.0 bp (SD = 856.63 bp), whereby the smallest mitogenome characterized so far, lacking control region but including the typical set of 37 genes, belongs to a bird body louse, Ciconiphilus decimfasciatus (14,102 bp) [50], and the largest, owing to a huge control region and without genome duplications, to the netspinning caddisfly, Potamyia horvati (27,450 bp) [51]. There are smaller and larger mitogenome versions in insects, but they represent either fragmented genomes, as found in the slender duck louse, Anaticola crassicornis (8,118 bp) [52], or whole genome duplications, as described for the Australian stingless bee, Tetragonula carbonaria (30,666 bp) [53]. Apart from these profound rearrangements, insect mitogenome size is rather conserved and length variation can be mostly attributed to the control region. As a note of caution, assemblies obtained with nanopore long-read technology with enough sequence depth compared with Illumina short-read mitogenome assemblies, have shown that the latter can potentially collapse repetitive elements, thus resulting in underestimated mitogenome sequenced lengths (e.g., [54]). With an average length of 16,442 bp (SD = 986.9 bp), beetle mitogenomes fall within the insect size range, but with less size variation: from the 14,476 bp of the cigarette beetle, Lasioderma serricorne [55], to the 22,714 bp in a European stag beetle, Lucanus cervus (Y. Ying, unpublished data). The mitogenomes of Taophila would be in the lowest 13.8% of this distribution, thus they can be classified as small insect mitogenomes, mainly owing to a relatively short origin of replication (857–1,021 bp). As stated above, most length variation of insect mitogenomes can be attributed to this non-coding region, and this is logically true for beetle mitogenomes as well, which can reach up to 5,000–6,000 bp in some species (e.g., [56, 57]). Also contributing to the small size of Taophila mitogenomes, it is worth mentioning the limited and reduced intergenic spacers (for a total of 47 bp in T. bituberculata and T. subsericea and 59 bp in T. mantillerii) and some gene overlap (0.614% in T. bituberculata, 0.705% in T. mantillerii, and 0.716% in T. subsericea, of total genome size).
The structure, gene order and orientation of these new mitogenomes is identical to previously published mitogenomes of other eumolpines and their closest relatives, the cryptocephalines and lamprosomatines [17, 18, 27]. Any differences detected were confirmed to be due to tRNA or rRNA annotation mistakes in published mitogenomes, and included: (1) swapped order of trnR and trnA, and inclusion of D-loop in the rrnS in Chrysochares punctatus [23]; (2) inversion of trnP in Colasposoma sp. [24]; (3) inversion of rrnS in Chrysodinopsis sp. [24]; and (4) incorrect naming of the rRNA genes in Chalcophana sp.,* Chlamisus sp., Chrysochus chinensis*,* Cryptocephalus flavolimbatus*,* Lachnaia urticarum*,* Physosmaragdina sp., Platycorynus sp., Spilopyra sumptuosa* and Syneta adamsi [17]. It is important to note that, even if gene identifications are correct for the cluster of tRNA genes between nad3 and nad5 genes in Nie et al. [17], both their Figs. 1 and 2 show trnE (Glutamate) inverted in the clade of Cryptocephalinae, Lamprosomatinae and Eumolpinae. Expectedly, Taophila mitogenomes include the novel order of the tRNA cluster between nad3 and nad5 that is synapomorphic for this clade [17]. Other than that, the characteristics of these new mitogenomes are the same as found in most Coleoptera and consistent with the inferred characteristics of the ancestral insect mitogenome [58]. From the point of view of diversity of tRNAs, Taophila mitogenomes show the same pattern as described for most Coleoptera and insects [6, 59, 60], presenting 22 tRNAs for the 20 amino acids, whereby the redundant trnL and trnS recognize the first and third most used codons in the Taophila mitogenomes. Most initiation and stop codons in Taophila are also typical of insect mitogenomes, including some incomplete T- or TA- termination codons completed to functional TAA stop codons by postranscriptional polyadenylation [61]. Interestingly, some of the Taophila NADH dehydrogenase subunit genes can start with the unusual TTG (Leu) initiation codon, first reported from insects in a springtail [62], and later found in other insect mitogenomes, but nonetheless present at very low frequences and only for a few genes [63]. Codon usage proportions in the mitogenomes of Taophila are very similar to those deduced from other leaf beetles, with minor differences. For example, Arginine seems underutilized compared to Gastrolina chrysomelines [64], or Valine more frequently used when compared to Luperomorpha flea beetles [65], but the general pattern of amino acid use is highly conserved. Finally, RSCU analysis reveals a bias towards codons ending in A or T, another common feature of insect mitogenomes (e.g [66]). This bias is also reflected in the effective number of codons per gene, indicative of a subset of codons used preferentially, i.e., with A or T at third codon positions. In combination with RSCU data, the significant correlation between GC-content at third versus first and second codon positions (p-value = 0.0056) suggests uniform base use in these mitogenomes. Codon usage bias can thus be interpreted as predominantly due to mutation pressure rather than selection [42, 67]. Neutrality plot analysis is a common way to assess the alternative roles of these two forces acting on codon usage bias, with empirical results from different groups of insects sometimes supporting mutational bias, as it occurs in Taophila (e.g., [68, 69]), and sometimes the alternative of selection (e.g., [70, 71]).
Mitophylogenomics of eumolpines and relatives reveals phylogenetic relationships generally consistent with the current systematics of these groups [17, 27], where most nodes (82.9%) receive strong bootstrap support (BS = 95–100%), and all but four nodes the highest posterior probabilities (PP = 0.98–1.00.98.00). Nodes which lack support (BS = 52–73%) from mitogenome data also failed to be resolved in studies with denser taxonomic sampling and data from several single-copy nuclear protein-coding genes, suggesting that they could reflect “hard” polytomies, i.e., rapid diversification of those clades rather than a polytomy due to conflict or lack of resolving data [72, 73]. These polytomies include the relationships among the tribes in Cryptocephalinae (although Bayesian inference also lent some support for example to a closer relationship between Clytrini and Cryptocephalini; [27]), and also among evolutionary lineages in Eumolpini, one of which is the South Pacific lineage represented by Taophila [28]. More problematic, in this case, is the apparent lack of monophyly for genera represented by more than one species in the Eumolpinae clade, or the placement of some species (such as Chrysodinopsis sp.). However, we suggest that these unexpected placements are not due to low resolving power of mitogenomes to solve a systematic problem, but due to the deficient taxonomy in most groups of Eumolpinae today [74]. Two intimately related problems possibly produce the seemingly scrambled taxonomy found in our mitochondrial phylogenetic hypothesis. First, the uncertain underlying taxonomy and lack of modern revisions of the huge genera Basilepta,* Colasposoma* and Pseudocolaspis, do not exclude that they may be indeed paraphyletic groups. Secondly, the lack of taxonomic expertise in Eumolpinae hampers sound taxonomic identifications in many cases. Both factors greatly reduce our confidence in the identifications attached to some sequences, and those characterized by Song et al. [24] are particularly problematic. Samples from this study generate all the conflicts observed at the generic level in the tree (despite these samples clearly having correct tribal identifications). Unfortunately, these sequences lack public metadata that could help in tracking their origin, and represent Afrotropical and New World taxa that cannot be reliably identified with the Chinese faunistic references used by the authors [24].
Mitogenomes continue to offer ample advantages for their implementation in evolutionary studies, including practical issues such as the relative ease in their sequencing with NGS methodologies and annotation via dedicated analytical pipelines [4, 45]. The deep knowledge about the structure, function and dynamics of this molecule, also helps exploiting mitogenomic information, as well as spotting interesting peculiarities worth further investigation, such as gene rearrangements [4]. Increasing the taxonomic quality and density of mitogenome data, especially targeting evolutionary lineages not represented in the available mitogenomes, will benefit the formulation of phylogenetic hypotheses useful for systematic and other evolutionary studies. This trend and the addition of Taophila to the mitogenome pool will be crucial to eventually resolve the phylogenetic position of South Pacific Eumolpinae. This lineage, currently lacking known closest relatives, may prove highly relevant to incorporate Antarctica to biogeographical models of the Southern hemisphere, as suggested for other Chrysomelidae, such as Chilean Mylassini cryptocephalines [27]. Alternatively, the steady addition of mitogenomes from pooled or environmental samples (e.g., [75, 76]), may help revealing, if by chance, an extant missing link of South Pacific Eumolpinae and support a more conventional biogeographic hypothesis. But in this respect, it is important that the publication of new mitogenomic resources is supported by robust identifications and include taxonomic details of analyzed samples, such as metadata to allow not only comparisons derived from these data, but also tracing the origin of samples for their reassessment (e.g., [77, 78]).
Supplementary Information
Supplementary Material 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. 2023. https://www.R-project.org/