Molecular prevalence of Chlamydia spp. in wild birds from Qinghai Lake, China
Xiaomin Wu, Fan Lei, Yaqian Niu, Jiali Yu, Chao Chen, Te Ba, Lin Liang

TL;DR
This study found a high prevalence of Chlamydia in wild birds at Qinghai Lake, China, with evidence of potential cross-species transmission between birds and yaks.
Contribution
The study identifies cross-species transmission of Chlamydia abortus between wild birds and yaks in the Qinghai-Tibet Plateau.
Findings
Chlamydia spp. infected 28.8% of tested wild birds at Qinghai Lake.
C. abortus in birds showed 100% genetic identity with a strain from yaks, suggesting cross-species transmission.
C. avium and C. psittaci were also detected, highlighting zoonotic risks in the region.
Abstract
Chlamydia spp. are a group of gram-negative, obligate intracellular bacteria that represent significant pathogens causing chlamydiosis in both animals and humans. Avian chlamydiosis (AC), primarily caused by Chlamydia psittaci, C. avium, C. gallinacea, and C. ibidis, has been documented in over 460 avian species. Qinghai Lake, China’s largest saltwater lake and a critical overwintering site for migratory birds, served as the study area to investigate Chlamydia prevalence in wild birds. Fecal samples from 125 birds revealed an overall Chlamydia spp. infection rate of 28.8% (36/125), with three species identified: C. abortus (55.6%, 20/36), C. avium (44.4%, 16/36), and C. psittaci (13.9%, 5/36). Phylogenetic analysis through amplification of the 16 S rRNA (5 samples), IGS-23 S rRNA (6 samples), and ompA (5 samples) genes revealed that all sequences obtained in this study were assigned to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
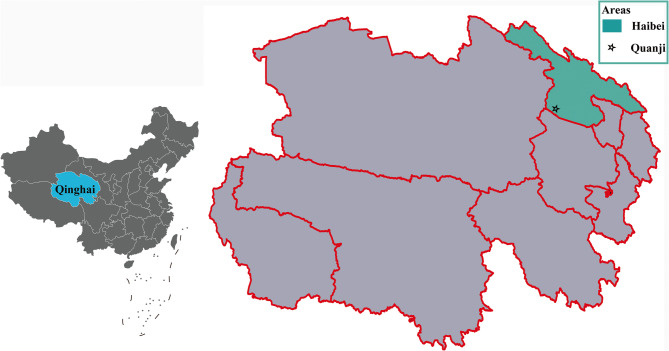 Figure 1
Figure 1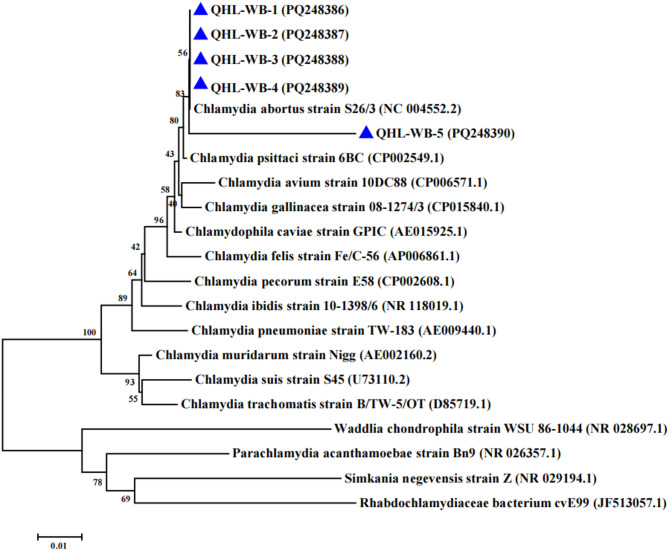 Figure 2
Figure 2 Figure 3
Figure 3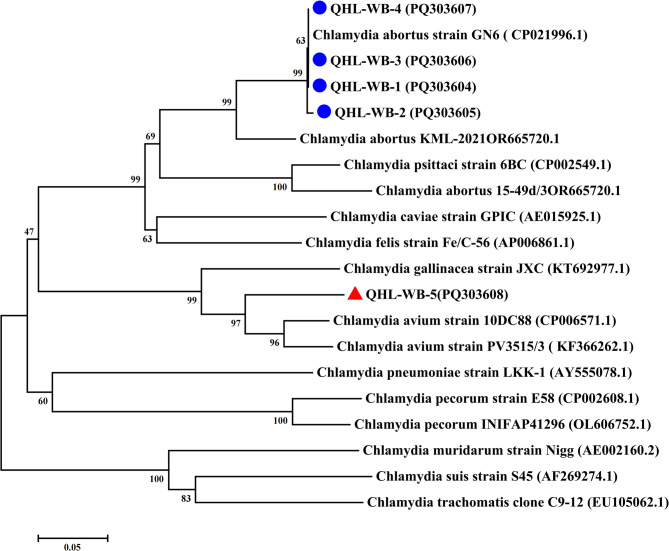 Figure 4
Figure 4 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsReproductive tract infections research · Gut microbiota and health · Bacteriophages and microbial interactions
Background
Chlamydia spp. are a group of gram-negative, obligate intracellular bacteria that causes chlamydiosis in animals and humans. Among these, C. psittaci, C. avium, C. gallinacea and C. ibidis are recognized as the causative agents of avian chlamydiosis (AC). C. psittaci is classified into 9 classical genotypes (A-F, E/B, WC, and M56) based on restriction fragment length polymorphism (RFLP) analysis of the ompA gene. Subsequent studies analyzing ompA sequences from wild birds have identified atypical genotypes, including 1 V, 6 N, Mat116, R54, YP84, and CPX0308 [1]. C. psittaci exhibits broad host specificity, infecting over 460 avian species [2]. Infected birds commonly present with respiratory distress, anorexia, emaciation, diarrhea, and, in severe cases, fatal outcomes. Additionally, C. psittaci poses significant zoonotic risks, threatening public health globally [3]. Human infections, often reported among poultry workers exposed to aerosols from contaminated feces or respiratory secretions, typically manifest as fever, cough, headache, and severe respiratory illnesses [4].
The global prevalence of C. psittaci in avian populations is notably high. Epidemiological surveys across 26 countries on five continents report an average infection rate of 19.5% [5]. Documented cases span diverse regions, including Egypt, Argentina, the Netherlands, and New Zealand, where varying infection rates have been observed in domestic birds [4, 6–8]. Notably, C. psittaci has also been detected in non-avian hosts such as horses and cats [9, 10]. In China, multiple studies highlight its widespread prevalence: a 31.09% seropositivity rate in northwest-region pigeons [11], a 26.2% positivity rate (602/2300) in poultry across 24 provinces [12], a 22.9% infection rate in cattle from 11 provinces [13], and a 34.2% detection rate in Taiwanese waterfowl farms [14]. Furthermore, C. psittaci was identified in 20 of 99 fecal samples from crested ibis [15], underscoring its pervasive presence in Chinese livestock and poultry.
Beyond C. psittaci, emerging species such as C. buteonis, C. ibidis, C. gallinacea, and C. avium have been linked to avian conjunctivitis and respiratory ailments. C. avium, primarily reported in European pigeons and parrots, was detected in 36.6% and 20.0% of pigeon droppings from Utrecht and Haarlem, respectively [8]. In China, surveys of chickens, ducks, geese, and pigeons revealed C. gallinacea, C. psittaci, C. suis, C. muridarum, and C. pecorum as predominant species, with C. gallinacea and C. psittaci occurring across all four avian hosts. Notably, no C. avium strains were identified in these studies [12].
Qinghai Lake, the largest saltwater lake in China, supports rich biodiversity and serves as a critical habitat for migratory waterbirds, including black-necked cranes (Grus nigricollis), pallas’s gull (Larus ichthyaetus), bar-headed geese (Anser indicus), brown-headed gulls (Chroicocephalus brunnicephalus), common cormorants (Phalacrocorax carbo), and whooper swans (Cygnus cygnus). Annually, approximately 4,000 whooper swans overwinter in this region. Despite its ecological significance, no data are available on Chlamydia prevalence in Qinghai Lake’s wild bird populations. This study aims to characterize Chlamydia species diversity through fecal sampling in the area. By enhancing diagnostic capabilities and informing disease control strategies, the findings will provide a scientific foundation for mitigating Chlamydia-related zoonotic risks, thereby offering significant public health and ecological applications.
Materials and methods
Study site and sample collection
Sampling was conducted at Qinghai Lake Nature Reserve, located in Quanji Township, Gangcha County, Haibei Tibetan Autonomous Prefecture, Qinghai Province, situated on the northwestern shore of Qinghai Lake. It forms an integral part of the Qinghai Lake National Nature Reserve. The coordinates 99°58′E and 37°23′N (Fig. 1), with an elevation of approximately 3,200 m above sea level. The wetlands surrounding Quanji Township serve as critical breeding and stopover grounds for rare waterbirds such as the bar-headed goose, whooper swan, and black-necked crane.
Fig. 1A map of China showing Qinghai Province and the geographical regions where wild bird feces were collected
Sampling was conducted at three distinct intervals: on October 1–3, October 12–14, and October 23–25 in 2023. A total of 125 fecal samples were collected from wild birds. The fecal samples encompassed 3 different avian species, including bar-headed goose, pallas’s gull, and brown-headed gull. Fecal samples were collected using sterile cotton-tipped swabs that were immediately placed into 2 mL sterile centrifuge tube. Species identification of the source birds was determined by a combination of factors: (1) sampling was conducted in specific areas habitually used and dominated by one of the three target species (bar-headed goose, pallas’s gull, or brown-headed gull), and (2) immediate collection of fresh feces after the birds were observed to leave the spot, with identification confirmed by the extensive field experience of the reserve staff in recognizing avian droppings. While this is a standard non-invasive method for field ecology studies, we acknowledge its potential limitations. Ethical approval was not required for this observational study, as samples were non-invasively collected by reserve staff during routine sanitation procedures. To preserve Chlamydia viability and diversity, fresh fecal materials were prioritized during collection. All samples were immediately frozen at − 80 °C until processing.
DNA extraction
Fecal swabs were immersed in 1 mL phosphate-buffered saline (PBS), vortexed for 10 min, and thoroughly squeezed to remove residual liquid. Genomic DNA was extracted using the TIANamp Genomic DNA Kit (TIANGEN, China), eluted in 120 µL of elution buffer, and stored at − 20 °C until use.
Real-time PCR assays to identify Chlamydiaceae species and DNA sequencing
All DNA samples (n = 125) underwent preliminary screening with a Chlamydiaceae-specific 23 S rRNA-targeting qPCR assay as described by Ehricht et al. [16]. Chlamydiaceae-positive samples were subsequently tested in duplicate using species-specific qPCR protocols for C. pecorum [17], C. abortus [17], C. psittaci [17], C.avium [18], C. gallinacea [18], and C. pneumoniae [19]. Following Mitura et al.‘s criteria [20], a cycle threshold (Ct) cut-off value of 38.0 was applied, corresponding to the assay’s lower detection limit. Positive DNA samples were further confirmed by amplification and sequencing of specific fragments of ompA, 16 S rRNA, and 16 S rRNA-23 S rRNA intergenic spacer together with 23 S rRNA domain I (IGS-23 S rRNA) as described previously [21–23]. PCR products were sequenced (Tsingke Biotechnology Co., Ltd., Xi’an, China). The obtained sequences were deposited in the GenBank database with the following accession numbers.
Phylogenetic analysis
Five 16 S rRNA sequences, six IGS-23 S rRNA sequences, and five ompA sequences (GenBank: PQ248390–PQ248396, PQ276123–PQ276128, PQ303604–PQ303608) were analyzed using MEGA 7.0. Sequence homology was verified via BLASTn against NCBI databases, followed by multiple alignment with reference Chlamydia strains. Phylogenetic trees for 16 S rRNA (1,400 bp), IGS-23 S rRNA (1,000 bp), and ompA (1,050 bp) were reconstructed using the neighbor-joining method with 1,000 bootstrap replicates under the Maximum Composite Likelihood model.
Results
By screening 125 wild bird fecal samples, Chlamydia spp. were detected in a total of 36 fecal samples with an overall prevalence of 28.8% (36/125). Three species were identified: C. abortus (55.6%, 20/36), C. avium (44.4%, 16/36), and C. psittaci (13.9%, 5/36), while C. pecorum, C. gallinacea, and C. pneumoniae were undetected (Table 1). The distribution of these pathogens across the thress bird species is detailed in Table 2. As shown, C.abortus was the most frequently detected species across all host birds. To further confirm the presence of the Chlamydia spp., 16 S rRNA (5 samples), IGS-23 S rRNA (6 samples), and ompA (5 samples) gene amplicons were successfully amplified and sequenced from high-concentration positive samples. Comparative phylogenetic analysis demonstrated that all sequences were assigned to the Chlamydiaceae family. C. abortus and C. avium identified in this study were all closely related to their respective reference sequences available in GenBank and altogether showed a similar topology (Figs. 2, 3 and 4). The ompA sequence of C. abortus obtained in this study clustered closely with the reference strain GN6 (CP021996.1) isolated from aborted yak fetuses, showing 100% sequence identity, while C. aviumshowed high genetic diversity, and sharing 87.40% identity with C. avium reference strain 10DC88 (Figure S1).Table 1. Detection of Chlamydia in bird feces in Qinghai lake areaDistrictSample typeChlamydia positive sample/sample numberNumber of positive samples for Chlamydia species/Number of positive samples for Chlamydia familyChlamydia abortusChlamydia aviumChlamydia psittaciQinghai Lakefeces36/12820/3616/365/36Table 2Prevalence of Chlamydia species in wild bird species from Qinghai lakeBird SpeciesChlamydia positive samples/no. of samplesNo of Chlamydia spp positive samplesC.abortusC.aviumC.psittaciBar-headed goose (Anser indicus)15/521072Pallas’s gull(Larus ichthyaetus)12/38552Brown-headed gull(Chroicocephalus brunnicephalus)9/35541Total36/12520165Co-infections with multiple Chlamydia species were detected in some samples, therefore the sum of the individual species counts may exceed the total number of positive samplesFig. 2Phylogenetic tree based on the 16S rRNA gene fragment (about 1400 bp) of Chlamydia. The phylogenetic tree includes representative sequences of established Chlamydiaceae species sequences as well as sequences discovered in this study (GenBank accession number was marked by blue triangle) were included Parachlamydia acanthamoebae strain Bn9, Rhabdochlamydiaceae bacterium cvE99, Simkania negevensis strain Z and Waddlia chondrophila strain WSU 86–1044 were used as an outgroup. Based on these alignments, phylogenetic trees were constructed by the neighbour-joining method using the Maximum Composite Likelihood model with MEGA 7.0.Fig. 3. Phylogenetic tree based on the 16S-23S intergenic spacer and full length of 23S rRNA domain I fragment (about 910 bp) of Chlamydia. The phylogenetic tree includes representative sequences of established Chlamydiaceae species sequences as well as sequences discovered in this study (GenBank accession number was marked by blue circles and red triangles) were included Candidatus Rhabdochlamydia sp. W744 and Simkania negevensis strain Z were used as an outgroup. Based on these alignments, phylogenetic trees were constructed by the neighbour-joining method using the Maximum Composite Likelihood model with MEGA 7.0.Fig. 4. Phylogenetic tree based on the ompA gene fragment (about 950 bp) of Chlamydia. The phylogenetic tree includes representative sequences of established Chlamydiaceae species sequences as well as sequences discovered in this study (GenBank accession number was marked by blue circles and red triangles). Based on these alignments, phylogenetic trees were constructed by the neighbour-joining method using the Maximum Composite Likelihood model with MEGA 7.0.
Discussion
This study investigating Chlamydiaceae prevalence in wild birds of Qinghai Lake, China, revealed an overall infection rate of 28.8% (36/125). Three species were identified: C. abortus (55.6%, 20/36), C. avium (44.4%, 16/36), and C. psittaci (13.9%, 5/36). Notably, this constitutes the inaugural documentation of Chlamydiaceae in Qinghai Lake’s avian populations and highlights C. abortus as a novel avian pathogen in China. Our findings align with domestic poultry surveillance data from China (26.2%, 602/2,300) [12], yet exceed rates reported in Brazilian backyard chickens (25.2%, 60/238) [24], Swiss turkeys (0.7%, 7/1008) [25], and endangered Crested Ibis populations (20.2%, 20/99) [15]. Discrepancies may arise from variations in host susceptibility, sampling strategies (e.g., fecal vs. cloacal swabs), and diagnostic methodologies (qPCR vs. ELISA).
Although AC caused by C. psittaci and emerging species like C. avium is typically subclinical but occasionally fatal [26–28]. While our data revealed a relatively low C. psittaci prevalence (13.9%) compared to regional poultry studies. Specifically, prevalence rates in Changsha poultry (27.5%) [29], Hainan domestic geese (25.6%) [30], Lanzhou pigeons (31.09%), Weifang and Beijing cities parrots (35.37%) [11, 31], exceeded our observations, potentially due to higher host density. Conversely, our rates surpassed those reported in Swedish garden birds (2.2%), Brazil backyard chickens (0.0%), Sichuan and Shandong birds (3.13%), and Hong Kong pet birds (0.97%) [15, 24, 32, 33]. This difference may be related to the ecological niche differences between wild and captive populations. These comparative analyses underscore the importance of host species ecology in chlamydial epidemiology.
In this study, our data showed that C. abortus was the dominant species in wild birds in the Qinghai Lake, representing 55.6% (20/36) of the Chlamydiaceae-positive sample. As far as we know, there are no previous reports of C. abortus in birds in China. While C. abortus has been sporadically reported in Argentinian psittacines [34], its detection in Chinese wild birds constitutes a novel finding. To further characterize the C. abortus, phylogenetic analysis of ompA sequences (PQ303604–PQ303606) revealed that C. abortus ompA sequences were all closely related to their respective reference sequences available in GenBank and altogether showed a similar topology. 100% identity with C. abortus reference strain GN6 (CP021996.1) by nucleic acid comparison, originally isolated from aborted yak fetuses on the Qinghai-Tibetan Plateau [35]. This genomic congruence suggests potential cross-species transmission pathways: wild birds may act as reservoir hosts, contaminating shared aquatic environments through fecal shedding, thereby exposing sympatric yak populations to infection risks. Subsequent human infections could occur through direct livestock contact or aerosol exposure during pastoral activities. Such interspecies transmission dynamics could lead to wildlife population declines through reproductive impairments and ecosystem destabilization, necessitating comprehensive surveillance programs.
Our study also has some limitations. One is the modest sample size may limit the statistical power to fully assess the diversity and distribution of Chlamydia species in this ecosystem. Expanding the sample size in future studies would be valuable to confirm these initial insights and better understand transmission dynamics. Another is the exclusive use of randomly collected fecal samples introduces a potential overestimation risk, as multiple samples might originate from individual birds. Furthermore, the hypothesized cross-species transmission of C. abortus between wild birds and livestock, while supported by genetic identity, would be more conclusively demonstrated by concurrent testing of sympatric livestock in the same region. Future longitudinal studies should include such sampling to provide direct evidence of pathogen circulation across species boundaries. Finally, technical constraints affected molecular characterization: despite 36 Chlamydia-positive samples, successful amplification of 16 S rRNA, IGS-23 S rRNA, and ompA genes was only achieved in 5–6 cases due to low bacterial DNA loads. The reliance on qPCR without confirmatory sequencing for C. psittaci detection represents an additional constraint. Future investigations should incorporate multianatomic sampling (cloacal/oropharyngeal swabs) and meta-genomic approaches to enhance detection sensitivity and phylogenetic resolution.
Conclusion
To summarize, this study illustrates that C. abortus, C. avium, and C. psittaci were detected in the wild birds in Qinghai Lake, China. C. abortus was the dominant species in wild bird in Qinghai Lake. Sequences of C. abortus obtained in this study were 100% identity with that of the C. abortus reference strain GN6 (CP021996.1). Furthermore, it may contribute to understanding the Chlamydia diversity and how it is transmitted.
Supplementary Information
Supplementary Material 1. Figure. S1. Alignment of the two ompA sequences from C. avium reference strain (10DC88) and a field prevalent strain (QHL-WB-5) in the wild birds in China.