A novel deletion upstream of POU3F4 in a Chinese family with X-linked deafness 2 and a literature review
Yanting Yang, Shengwen Huang, Yao Liu, Dan Mu, Ting Bai, Xiaosha Jing, Lingling Xing, Hongqian Liu

TL;DR
A new genetic deletion upstream of the POU3F4 gene is found in a Chinese family with X-linked deafness 2, offering insights into the condition's genetic basis.
Contribution
Identification of a novel 795.5 kb deletion upstream of POU3F4 and its impact on gene expression in DFNX2.
Findings
A novel deletion of 795.5 kb upstream of POU3F4 was identified in a Chinese family with X-linked deafness 2.
The deletion caused reduced expression of POU3F4 and its downstream target genes in affected individuals.
This study expands the known spectrum of upstream deletions associated with DFNX2.
Abstract
X-linked deafness 2 (DFNX2) is a rare hereditary hearing loss characterized by progressive conductive and sensorineural hearing loss and a pathognomonic temporal bone deformity. DFNX2 is caused by mutations in the coding sequence or deletions upstream of POU3F4. Only 12 upstream deletions of POU3F4 associated with DFNX2 have been reported, and the precise mechanisms underlying its pathogenesis remain fully elucidated. Whole-genome Sequencing (WGS) and linkage analysis were performed to identify potential genetic etiologies. Gap-PCR and Sanger sequencing were used to validate candidate pathogenic variants and elucidate the breakpoints. Quantitative Polymerase Chain Reaction (qPCR) was conducted to evaluate the altered expression of both POU3F4 and its downstream target genes. Here, we identified a novel deletion approximately 795.5 kb in length, located about 140 kb upstream of POU3F4…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
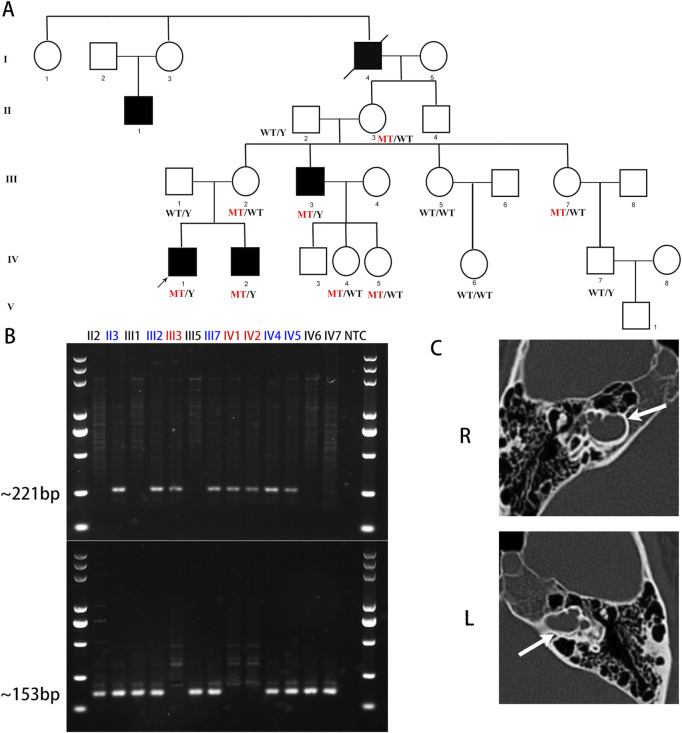 FIGURE 1
FIGURE 1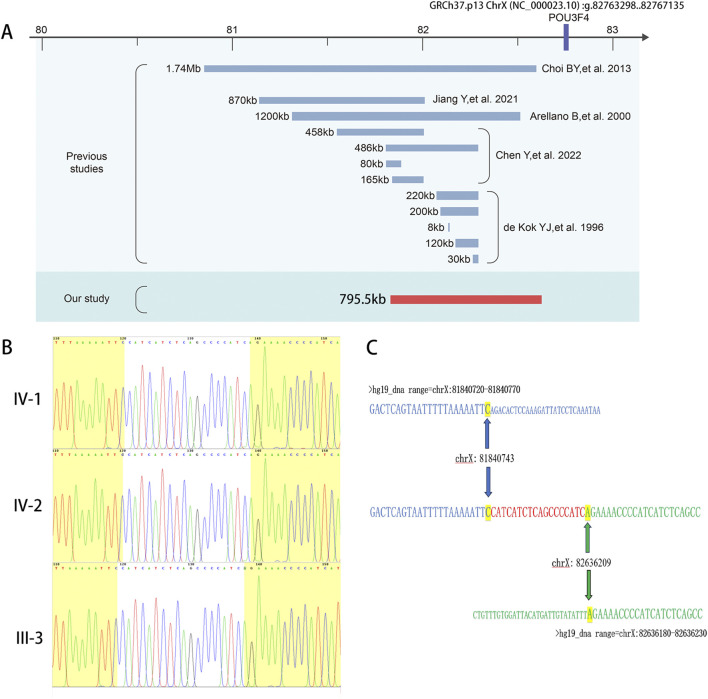 FIGURE 2
FIGURE 2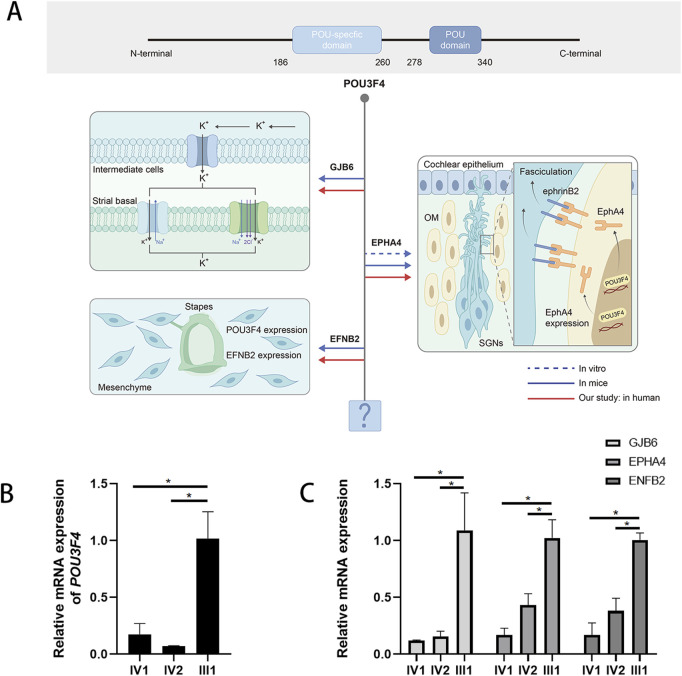 FIGURE 3
FIGURE 3| Sequence information | Clinical information | Ref | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genomic position | Length | Distance upstream POU3F4 | Ethnicity | Testing technique | Hearing loss | Malformations | Vestibular dysfunction | Other findings | |||
| Onset(m) | Severity | Type | |||||||||
| – | 220 kb DEL | NA | NA | Southern blot analysis | NA | NA | NA | NA | NA | – |
|
| – | 200 kb DEL | NA | NA | Southern blot analysis | NA | NA | NA | NA | NA | – | |
| – | 8 kb DEL | NA | NA | Southern blot analysis | NA | NA | NA | NA | NA | – | |
| – | 120 kb DEL | NA | NA | Southern blot analysis | NA | NA | NA | NA | NA | – | |
| – | 30 kb DEL | NA | NA | Southern blot analysis | NA | NA | NA | NA | NA | – | |
| – | 1,200 kb DEL | 250kb | Danish | dideoxy dye-terminator cycle sequencing | NA | profound | SNHL | IP-III | EVA | Epilepsy, hyperkinetic syndrome, and goiter |
|
| ChrX: g. 80851535_82597832 | 1.74Mb DEL | 165kb | Korean | aCGH | 13 | profound | Mixed | IP-III | NA | – |
|
| ChrX: g.81140313_82011100 | 870 kb DEL | 750kb | Chinese | Nanopore + Sanger | 6 | profound | SNHL | IP-III | NA | – | ( |
| ChrX: g.81548899_82006629 | 458 kb DEL | 1,502 kb | Chinese | Nanopore + Sanger | 5 | profound | NA | IP-III | – | Mild autism | ( |
| ChrX: g.81806051_82292259 | 486 kb DEL | 1,217 kb | Chinese | Nanopore + Sanger | 5 | profound | NA | IP-III | – | – | |
| ChrX: g.81807331_81887213 | 80 kb DEL | 876 kb | Chinese | Nanopore + Sanger | 5 | profound | NA | IP-III | – | Atrial septal defect | |
| ChrX: g.81839469_82004841 | 165 kb DEL | 1,503 kb | Chinese | Nanopore + Sanger | 4 | profound | NA | IP-III | – | – | |
| ChrX: g. 81840743_82636209 | 795.5 kb DEL | 140 kb | Chinese | NGS + Sanger | 3 | profound | Mixed | IP-III | – | Our study | |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHearing, Cochlea, Tinnitus, Genetics · RNA modifications and cancer · Congenital heart defects research
1 Introduction
DFNX2 (OMIM: #304400) accounts for approximately 50% of all families with X-linked nonsyndromic deafness, which only makes up 2%–3% of hereditary hearing loss (Petersen et al., 2008; Corvino et al., 2018). In 1971, DFNX2 was first described as an X-linked condition characterized in males by profound mixed deafness, vestibular abnormalities, and congenital fixation of the stapes (Nance et al., 1971). Subsequently, in 2002, the cochlear abnormality associated with DFNX2 was classified as incomplete partition type III (IP-III) (Sennaroglu and Saatci, 2002).
The POU domain, class III, transcriptional factor 4, POU3F4 (NM_000307.5), is located on chromosome Xq21 and encodes a transcription factor belonging to the POU-domain family, which includes a POU-specific domain and a POU-homeodomain (Mathis et al., 1992). In 1995, POU3F4, also known as BRN4, was identified as the gene responsible for DFNX2 (de Kok et al., 1995). The upstream deletions have been identified as a causative factor for DFNX2. The first report of a deletion upstream of POU3F4 in patients with X-linked mixed deafness dates back to 1971 (Nance et al., 1971). In 1996, deletions occurring 900 kb upstream of POU3F4 were identified as a hotspot for DFNX2 (de Kok et al., 1996). However, only 12 upstream deletions of POU3F4 associated with DFNX2 have been reported (de Kok et al., 1996; Arellano et al., 2000; Choi et al., 2013; Jiang et al., 2021; Chen et al., 2022). And the details of the underlying pathogenesis associated with these deletions remain unclear (Chen et al., 2022). Although the role of POU3F4 in the inner ear is indispensable, no transcriptional targets of POU3F4 have been thoroughly investigated. It still leaves an unresolved question regarding the functional network downstream of this transcription factor (Bernardinelli et al., 2023).
In this study, we identified a novel deletion situated upstream of POU3F4 in a large Han family in China. Furthermore, we investigated the reduced expression of POU3F4 and the downregulation of its downstream target genes in patients with this upstream deletion, a finding that has not been reported in prior studies.
2 Materials and methods
2.1 Study participants
The Chinese family involved in this study was recruited from the Medical Genetics/Prenatal Diagnosis Centre of West China Second University Hospital, Sichuan University, Chengdu, China. The family consists of 26 members, with 5 affected individuals (all male). These patients exhibited profound congenital hearing loss and underwent comprehensive otological examinations and systematic assessments, including pure-tone audiometry and radiological examinations, in the local Department of Otolaryngology, Head and Neck Surgery (ENT Department). They sought genetic analysis to identify the underlying causes of their hearing impairment as the new generation reached reproductive age. Family history was collected by physicians from the Genetic Consulting Center. This study received ethical approval from the Institutional Review Board of West China Second University Hospital, Sichuan University. Informed consent was obtained from each participant or, for minors, their legal guardians. And the study was conducted in accordance with the Helsinki Declaration.
2.2 Genetic studies
We conducted Whole-genome Sequencing (WGS) using DNA obtained from patient samples as follows. Genomic DNA was extracted from peripheral blood samples utilizing the FitAmp Plasma/Serum DNA Isolation Kit (P-1004-1, Epigentek, Farmingdale, New York, United States). Sequencing was carried out on the Illumina HiSeq X system (Illumina, San Diego, California, United States). Additionally, we performed genetic linkage analysis as described in a previous study (Smith et al., 2011).
The PCR reaction was conducted using Golden Star T6 Super PCR Mix (TSE101, TSINGKE, Beijing, China) in a thermal cycler, beginning with an initial denaturation step at 98 °C for 1 min, followed by 34 cycles of denaturation at 98 °C for 1 min, annealing at 60 °C for 15 s, and extension at 72 °C for 1 min. After thermal cycling, a final extension was performed at 72 °C for 1 min, after which the samples were immediately placed on ice. PCR amplification was carried out using the ProFlex PCR System (Thermo Fisher Scientific, Waltham, Massachusetts, United States). Sequencing of the PCR products was performed on an ABI377A DNA sequencer (Applied Biosystems, Foster City, California, United States). We designed deletion-specific primers and wild-type (WT) specific primers for the amplification of the identified 795.5 kb deletion. Primers POU3F4-DEL-F and POU3F4-DEL-R were located upstream and downstream of the deletion breakpoint, respectively, while primers POU3F4-WT-F and POU3F4-WT-R were designed to be located in the deletion region. A complete list of the deletion-specific primer pairs is provided in Table 1. The PCR products were then resolved by gel electrophoresis and scored as either present or absent. Sanger sequencing was employed to elucidate the breakpoints of the deletion.
2.3 RNA extraction and quantitative real-time PCR (qPCR)
The RNA from the blood sample was extracted using Trizol (15596018, Thermo Fisher Scientific). Specifically, 1 mL of Trizol was used to solubilize the blood sample for 5 min at room temperature, followed by the addition of 0.2 mL of chloroform to facilitate phase separation for 2–3 min at room temperature. After centrifugation at 10,000 g for 10 min, the upper clear phase was mixed with 0.5 mL of isopropanol to precipitate the RNA for 10 min. The precipitated RNA was then collected by centrifugation at 10,000 g for 10 min at 4 °C. The RNA was re-extracted with phenol after resuspension and subsequently re-precipitated with ethanol. The concentration and purity of the RNA were assessed using a NanoDrop 2000 (Thermo Fisher Scientific).
We performed reverse-transcription to obtain cDNA, and qPCR was accomplished using KAPA SYBR^®^ FAST (suitable for qPCR, 2 ×, Universal) (KK4602, Roche, Basel, Switzerland) in an Applied Biosystems™ 7,500 Real-Time PCR System (Applied Biosystems). The PCR conditions were 40 cycles of denaturation at 95 °C for 5 s and annealing at 60 °C for 30 s. We analyzed results using the 2^−ΔΔCT^ method, with the expression of GAPDH serving as a reference gene (Livak and Schmittgen, 2001). Each reaction was repeated three times. The primers for quantitative real-time PCR (qPCR) are displayed in the list of primer pairs (Supplementary Table S1).
2.4 Statistical analysis
All data are expressed as arithmetic means ± SEM and analyzed with GraphPad Prism software (version 8.3.0, GraphPad Software, San Diego, California, United States). Significant differences between data sets were verified by a nonparametric test, as appropriate. A p-value <0.05 was considered statistically significant; (n) corresponds to the number of independent measurements.
3 Results
3.1 Characteristics of the clinical phenotype
IV-1 and IV-2 were diagnosed with profound bilateral mixed hearing loss at the ENT department. IV-1, now 26 years old, received his hearing aids at the age of 3. Despite being able to engage in social conversations without experiencing tinnitus or vestibular symptoms, his speech and communication skills remain impaired. IV-2, currently 16 years old, received hearing aids at the age of 6. With the assistance of speech and language therapy, IV-2 has shown significant improvement in his speech and communication abilities. He now attends a mainstream high school, excels academically, and denies experiencing tinnitus or vestibular symptoms. At the age of 15, his speech test results in both quiet and noisy environments were deemed satisfactory. Both IV-1 and IV-2 underwent presurgical evaluations, including pure-tone audiometry and radiological examinations. However, due to the considerable time elapsed since their treatment, complete clinical data are no longer accessible. Only the temporal bone CT scans of IV-1 were obtained. The radiological images demonstrate symmetrical cochlear hypoplasia and abnormal communication between the bottom of the internal auditory canal and the vestibule, which are characteristic of DFNX2 (Figure 1C). These characteristics are all consistent with the previously reported probands harboring deletions upstream of POU3F4 (Table 1). In previous reports, one subject was described by Arellano et al. as having bilateral vestibular areflexia (Arellano et al., 2000), and one subject was described by Chen et al. as having mild autism (Chen et al., 2022) (Table 1). However, neither IV-1 nor IV-2 reported any vestibular dysfunction or manifestations related to the autism spectrum.
A novel deletion upstream of POU3F4 was identified in the DFNX2 family. (A) The pedigree of this DFNX2 family is presented. The black squares represent affected individuals with DFNX2 and a hemizygous deletion, upstream of POU3F4. (B) The amplicons generated by deletion-specific primers measured 221 bp, whereas the amplicons from wild-type (WT)-specific primers measured 153 bp. Individuals with a hemizygous deletion are indicated in red, while carriers with a heterozygous deletion are in blue. (C) Temporal bone CT scans of the patient (IV-1) revealed dilation of the internal auditory canal, with communication observed between the lateral canal and the basal cochlear turn. The white arrows indicate the internal auditory canal.
After gathering information regarding the family history, we constructed a pedigree chart (Figure 1A). The chart illustrates additional affected males (II-1 and III-3) who also experience profound bilateral hearing loss. Adult females in the family denied any hearing impairments. Notably, individuals II-3 (80 years old) and III-2 (50 years old) assert having normal hearing but have declined additional testing. The family exhibits a classic X-linked inheritance pattern.
3.2 Identification of a novel deletion in the upstream of POU3F4
To ascertain a genetic diagnosis, we performed Trio whole-exome sequencing (Trio-WES) on the family members of the siblings (IV-1, IV-2, III-1, and III-2). The results were negative. Considering the evident X-linked inheritance pattern in the pedigree, we subsequently conducted Trio whole-genome sequencing (Trio-WGS) and genetic linkage analysis to explore potential candidate molecular diagnosis. Remarkably, we identified a 795.5 kb deletion located at GRCh37 (chrX): g. 81840740-82636210, approximately 140 kb upstream of the POU3F4 gene (Figure 2A), which is known to be the causative gene for DFNX2. The large deletion was absent from gnomAD, Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), or DECIPHER (https://decipher.sanger.ac.uk/browser).
Genetic analysis of this novel 795-kb deletion located upstream of the POU3F4 gene. (A) A schematic illustration of the upstream region of POU3F4 is presented, displaying the deletions identified in our study as well as those reported previously. (B) Sanger sequencing revealed 18-bp novel added nucleotides (CATCATCTCAGCCCCATC) in the breakpoint region. The nucleotides that matched the reference sequence are highlighted in yellow. (C) Alignment of the sequenced junctions with the reference genome sequence. Proximal and distal reference sequences are shown in green and blue, respectively. The junction is shown in red. Distal (top, blue) and proximal (bottom, black) sequences were aligned against the junction sequence (middle).
Currently, only 12 deletions upstream of POU3F4 have been described in previous reports (de Kok et al., 1996; Arellano et al., 2000; Choi et al., 2013; Jiang et al., 2021; Chen et al., 2022). We conducted a comprehensive review of the previously reported deletions upstream of POU3F4 and the identified deletions upstream of POU3F4 (Table 1). The deletions associated with DFNX2 exhibited variable sizes, ranging from 8 kb to 1.74 Mb, with approximate locations 140 kb–1,503 kb upstream of POU3F4 (Figure 2A). Notably, there is no common overlapping region among these deletions. Remarkably, our investigation revealed that the downstream breakpoint of the novel deletion we detected is the closest to the POU3F4 gene reported thus far. Our findings contribute to broadening the spectrum of pathogenic deletions associated with DFNX2, located upstream of POU3F4.
To assess the presence of a genomic deletion in the family, we performed polymerase chain reaction (PCR) analysis. Electrophoresis confirmed successful amplification of the junction sequence (Figure 1B). The male patients (III-3, IV-1, and IV-2) only showed the amplicons of deletion-specific primers (221bp) (Figure 1B). And tested female family members, II-3, III-2, III-7, IV-4, and IV-5, showed both the amplicons of deletion-specific primers (221bp) and amplicons of WT-specific primers (153bp) (Figure 1B). We consider these five persons as X-linked female carriers with this heterozygous deletion. We also researched the junction sequence with amplicons of deletion-specific primers by Sanger sequencing (Figure 2B). The results of the junction sequence indicated that the deletion is actually located at GRCh37. p13 ChrX (NC_000023.10): g. 81840743_82636209. Notably, the sequencing data revealed the presence of 18 novel nucleotides (CATCATCTCAGCCCCATC) added at the junction points (Figure 2C). This finding supports the hypothesis that this deletion may be the genetic cause of the observed phenotype in this family.
3.3 The 795.5 kb deletion upstream of the POU3F4 impairs its expression
To determine whether the deletion (NC_000023.10: g. 81840743_82636209del) located upstream of POU3F4 affects the mRNA expression of the POU3F4 gene, we conducted a quantitative real-time PCR (qPCR) assay using RNA extracted from the peripheral leukocytes of the patients. The relative expression levels of POU3F4 mRNA in affected siblings (IV-1 and IV-2) with the upstream deletion of POU3F4 that we detected were significantly lower compared to the male normal control (III-1) (Figure 3B).
*The deletion upstream of the POU3F4 we detected impairs the expression of POU3F4 and its downstream genes. (A) Partial mapping of the transcriptional target network of POU3F4: integrating findings from previous studies with our current research. (B) The result of qPCR shows a sharp decrease in the mRNA level of the affected brothers compared to their healthy father. Three independent experiments were performed. indicate the p-value <0.05 (nonparametric test; error bars, mean ± SEM). (C) The result of qPCR also shows that the relative expression levels of GJB6, EPHA4, and EFNB2 mRNA in the affected patients were significantly lower compared to normal people. Three independent experiments were performed. * indicate the p-value <0.05 (nonparametric test; error bars, mean ± SEM).
3.4 Upstream deletion also affected the expression of downstream targets of POU3F4
To gain a deeper understanding of the deletion identified upstream of POU3F4, we also assessed the mRNA expression levels of GJB6, EPHA4, and EFNB2(Ephrin-B2), which are known to be influenced by POU3F4 during inner ear development (Figure 3A). The relative expression levels of GJB6, EPHA4, and EFNB2 mRNA in the affected patients were also significantly lower compared to those of normal people (Figure 3C). According to previous studies, the impact of POU3F4 on downstream genes has been demonstrated in vitro and in mice (Figure 3A). Our findings further substantiate the effect of the deletion detected upstream of POU3F4 on the expression of its downstream genes. Additionally, the influence of POU3F4 on its downstream genes has been observed in humans with DNFX2 for the first time. Meanwhile, these findings also provide new evidence to investigate the functional network downstream of POU3F4.
Thus, the novel deletion (NC_000023.10: g. 81840743_82636209del) at approximately 140 kb upstream of the POU3F4 gene disrupts POU3F4 expression, leading to impaired function. This negative function further impacted other important proteins in inner development, potentially culminating in the DFNX2 phenotype. Collectively, these findings strongly indicate that the identified deletion upstream of POU3F4 could serve as the molecular basis for the hearing loss observed in this family.
4 Discussion
In a large Han family with DFXN2, we detected a novel 795.5 kb deletion on ChrX (NC_000023.10: g. 81840743_82636209), situated approximately 140 kb upstream of POU3F4. Following a negative result from whole-exome sequencing (WES), we conducted whole-genome sequencing (WGS) and genetic linkage mapping using SNP genotypes extracted from the WGS data to investigate candidate molecular diagnosis. In our study, we first demonstrated that deletion upstream of POU3F4 impacted the mRNA expression of POU3F4. Furthermore, the decreased RNA expression levels of downstream targets of POU3F4 were also first observed in patients with this deletion that we identified. Based on our experimental data, we propose that the 795.5 kb deletion upstream of POU3F4 may lead to DFNX2 by impairing the expression of both POU3F4 and its downstream functional network.
In our study, we also detected the presence of 18 novel nucleotides (CATCATCTCAGCCCCATC) inserted at the junction points. This finding is consistent with the previous report. In the study of Jiang Yi et al., non-homologous end joining (NHEJ), an important type of genome rearrangement (Ja et al., 2007), was identified as a mechanism for the deletion in the upstream region of POU3F4 (Jiang et al., 2021). Notably, NHEJ typically results in the loss of several nucleotides at each end of the DNA break (Hhy et al., 2016). The 18-bp sequence (CATCATCTCAGCCCCATC) we detected is the “information scar” at the joining points. It suggested that NHEJ may be the causal pathway for the novel 795.5 kb deletion located at approximately 140 kb upstream of POU3F4.
The POU3F4 gene, approximately 8,907 bp in length, is located on the X chromosome at the Xq21.1 locus and consists of a single coding exon that spans 1,506 bp (de Kok et al., 1995). In 1997, the first deletion upstream of POU3F4 in patients with X-linked mixed deafness was reported (Nance et al., 1971). Since then, 12 deletions upstream of POU3F4 associated with DFNX2 have been reported. These deletions exhibit different lengths and different distances from POU3F4. And there is no common overlap region among these deletions (Figure 2A). This phenomenon might mean that the underlying pathogenic mechanism is not as same as general copy number variants (CNVs). According to the previous study, the human POU3F4 gene is located in a 3-Mb gene desert region enriched in highly conserved non-coding regions (HCNRs), which are enriched in cis-regulatory elements (Alonso et al., 2009). For the tight regulation of gene expression in time and space in development, core promoters close to transcription start sites must interact with noncoding regulatory elements in their vicinity, termed enhancers (Robson et al., 2019). Enhancers and promoters communicate across large genomic distances through direct physical contact via chromatin folding, which is responsible for chromosome conformation (Robson et al., 2019). Deletions upstream of POU3F4 might affect chromosome conformation, thereby impact the function of cis-acting elements and contributing to the pathogenic mechanism. Further studies are needed to investigate the characterization and function of this region.
Meanwhile, the functional network downstream of POU3F4 has not been fully characterized so far (Bernardinelli et al., 2023). Loss of function of POU3F4 disrupts the assembly and localization of gap junction proteins, specifically connexin 26 (Cx26) and connexin 30 (Cx30), at the cell borders of cochlear supporting cells, thereby affecting the endolymphatic potential (EP) in the inner ear (Kidokoro et al., 2014). Additionally, Coate et al. demonstrated that the expression of EPHA4, a receptor tyrosine kinase, is decreased in Pou3f4 knockout (KO) mice, and that POU3F4 binds to the regulatory elements of EphA4 (Raft et al., 2014). EPHA4 is known to regulate spiral ganglion axon fasciculation, which is essential for appropriate auditory innervation (Raft et al., 2014). Furthermore, POU3F4 and the Eph receptor transmembrane ligand Ephrin-B2 exhibit a common spatiotemporal expression pattern during the organogenesis of the middle and inner ear, providing evidence for the role of POU3F4 in the development of the bony tissue surrounding the vestibular labyrinth (Raft et al., 2014). In our study, we directly confirmed the expression alteration of this transcriptional target by real-time PCR analysis in DNFX2 patients. Our findings not only confirm the pathogenicity of the identified deletion but also provide additional evidence for the further construction of downstream gene networks.
Previous reports on upstream deletions of POU3F4 lacked mRNA-level validation or verification. This limitation stemmed from the challenging nature of constructing animal or cell models for such deletions, which are situated in noncoding regions and range from 8 kb to 1.74 Mb in size. Despite the advancements in gene editing through CRISPR-Cas9, precise deletion of large DNA fragments, spanning from kilobases to megabases, remains a persistent challenge (Li et al., 2023). Choi et al. showcased the potential for accurate long genomic sequence deletions and replacements; however, they highlighted a relatively low deletion efficiency, with only about a 2% success rate observed for a 10 kb deletion (Choi et al., 2022). In humans, the POU3F4 gene is predominantly expressed in the nervous system. Nevertheless, mRNA of POU3F4 has also been detected in whole blood in the BioGPS database (BioGPS - your Gene Portal System). Quantitative real-time PCR is commonly employed in gene expression profiling (Zainal Ariffin et al., 2022). Hence, we utilized a qPCR assay with RNA extracted from peripheral leukocytes of patients to assess whether upstream deletion of POU3F4 impacts POU3F4 gene expression. Additionally, we performed qPCR to validate the expression of these downstream genes of POU3F4, further confirming the deleterious effect of the detected deletion. However, the specific pathogenic mechanism of the upstream deletion of the POU3F4 gene remains unclear and needs further study.
5 Conclusion
Our study identified a novel deletion located at approximately 140 kb upstream of POU3F4 in a large Han family with DFXN2. Our findings expand the spectrum of pathogenic deletions associated with DFNX2 that occur upstream of the POU3F4. We first revealed that the deletion upstream of POU3F4 impacted the mRNA expression of POU3F4 and its downstream genes in patients with this deletion. Our work presented more detailed information on the pathogenesis of deletion upstream of POU3F4 and provided further evidence to the understanding of downstream gene networks of POU3F4.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alonso M. E.Pernaute B.Crespo M.Gómez-Skarmeta J. L.Manzanares M. (2009). Understanding the regulatory genome. Int. J. Dev. Biol. 53, 1367–1378. 10.1387/ijdb.072428 ma 19247937 · doi ↗ · pubmed ↗
- 2Arellano B.Ramírez Camacho R.García Berrocal J. R.Villamar M.del Castillo I.Moreno F. (2000). Sensorineural hearing loss and Mondini dysplasia caused by a deletion at locus DFN 3. Arch. Otolaryngol. Head. Neck Surg. 126, 1065–1069. 10.1001/archotol.126.9.1065 10979118 · doi ↗ · pubmed ↗
- 3Bernardinelli E.Huber F.Roesch S.Dossena S. (2023). Clinical and molecular aspects associated with defects in the transcription factor POU 3F 4: a review. Biomedicines 11, 1695. 10.3390/biomedicines 11061695 37371790 PMC 10296620 · doi ↗ · pubmed ↗
- 4Chen Y.Qiu J.Wu Y.Jia H.Jiang Y.Jiang M. (2022). Genetic findings of sanger and nanopore single-molecule sequencing in patients with X-linked hearing loss and incomplete partition type III. Orphanet J. Rare Dis. 17, 65. 10.1186/s 13023-022-02235-7 35189936 PMC 8862311 · doi ↗ · pubmed ↗
- 5Choi B. Y.An Y.-H.Park J. H.Jang J. H.Chung H. C.Kim A.-R. (2013). Audiological and surgical evidence for the presence of a third window effect for the conductive hearing loss in DFNX 2 deafness irrespective of types of mutations. Eur. Arch. Otorhinolaryngol. 270, 3057–3062. 10.1007/s 00405-013-2386-3 23400403 · doi ↗ · pubmed ↗
- 6Choi J.Chen W.Suiter C. C.Lee C.Chardon F. M.Yang W. (2022). Precise genomic deletions using paired prime editing. Nat. Biotechnol. 40, 218–226. 10.1038/s 41587-021-01025-z 34650269 PMC 8847327 · doi ↗ · pubmed ↗
- 7Corvino V.Apisa P.Malesci R.Laria C.Auletta G.FranzéA. (2018). X-linked sensorineural hearing loss: a literature review. Curr. Genomics 19, 327–338. 10.2174/1389202919666171218163046 30065609 PMC 6030855 · doi ↗ · pubmed ↗
- 8de Kok Y. J.van der Maarel S. M.Bitner-Glindzicz M.Huber I.Monaco A. P.Malcolm S. (1995). Association between X-linked mixed deafness and mutations in the POU domain gene POU 3F 4. Science 267, 685–688. 10.1126/science.7839145 7839145 · doi ↗ · pubmed ↗