Deletions in (CCCT)n repeat regions belonging to the human pRNA gene inhibit its expression
Nikola Chmúrčiaková, Adam Nógell, Evgeny Smirnov, Dušan Cmarko

TL;DR
This study shows that deletions in specific DNA repeat regions reduce the expression of a gene important for ribosomal RNA production.
Contribution
The study identifies specific DNA sequence variations that inhibit pRNA gene expression and promote ribosomal RNA production.
Findings
SNVs and deletions in (CCCT)n repeats are more frequent in DNA than in RNA transcripts.
DNA variants inhibit pRNA expression and indirectly promote ribosomal RNA production.
(CCCT)n/(GGGA)n DNA repeats may form triplex structures that facilitate pRNA function.
Abstract
Ribosomal DNA (rDNA), the most abundantly expressed locus in the human genome, is represented by hundreds of units per cell. Each unit includes a 13 kb long region (47S rDNA) containing the genes of ribosomal particles, and a 30 kb long intergenic spacer (IGS). The 47S rDNA is transcribed with varying intensity in different units, some of which remain permanently silent. A key intermediator of this silencing is the promoter-associated RNA (pRNA) produced from a 2 kb long gene situated upstream of the rDNA transcription start site. Recent studies, including ours, suggest that the sequence variability, which normally occurs in mammalian cells, may account for the selective transcription of different rDNA units. The present work is based on the deep sequencing of a pRNA gene fragment and its RNA product and subsequent bioinformatic analysis. We found that a certain SNV, which converts the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
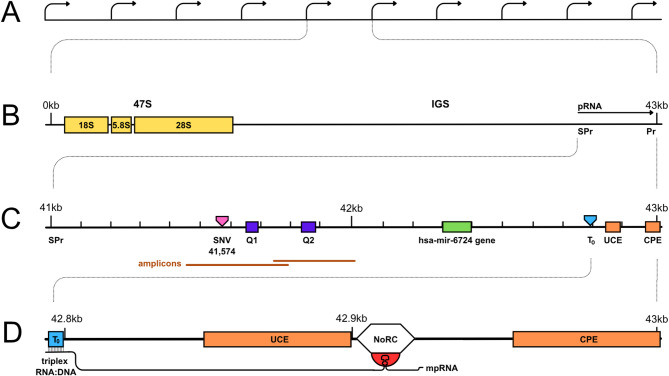 Figure 1
Figure 1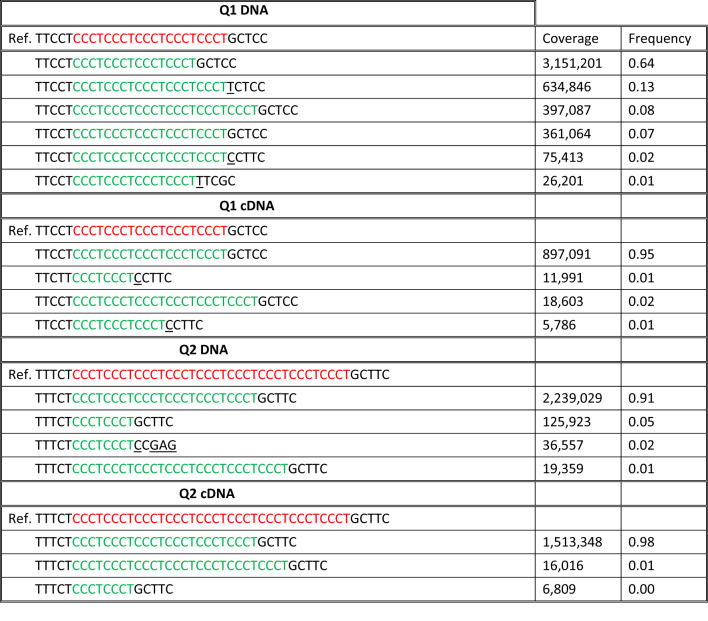 Figure 2
Figure 2- —https://doi.org/10.13039/100007397Univerzita Karlova v Praze
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA and protein synthesis mechanisms · RNA modifications and cancer · DNA and Nucleic Acid Chemistry
Background
Ribosomal DNA (rDNA), the most abundantly expressed locus in the human genome, is represented by hundreds of repeats or units per cell. Each unit consists of two parts: a 13 kb long 47S DNA containing the genes of ribosomal particles, and a 30 kb long intergenic spacer (IGS) (shown in Fig. 1A, B). The 47S DNA is transcribed by RNA polymerase I (pol I) with various intensity in different units, some of which remain permanently silent. The transcription is regulated, in the entire locus as well as in individual units, through multiple pathways targeting the state of pol I and its transcription factors or the chromatin structure of the promoter area [1–3].
Fig. 1. The structure of the human rDNA locus. A A cluster of the rDNA units. B Details of one unit including genes encoding 18S, 5.8S, and 28S ribosomal RNAs; intergenic spacer (IGS); rDNA promoter (Pr); spacer-promoter (SPr); the gene of promoter-associated RNA (pRNA). C The pRNA locus which includes the upstream control element (UCE) and the core promotor element (CPE) of the rDNA promoter; SNV_41,574_ (CCC > CCT) significantly correlating with DNA methylation status; regions of simple tandem repeats (CCCT)n marked Q1 and Q2; promoter-proximal terminator (T_0_); the gene encoding hsa-mir-6724. D Interaction of the mature pRNA (mpRNA) with T_0_, by forming RNA:DNA triplex, and with the nucleolar remodelling complex (NoRC), by forming a specific hair-pin structure. In this conception, no role is assigned to the sequence lying between SPr and T_0_
The promoter-associated RNA (pRNA) is an important inhibitor of the rDNA transcription [4–6]. In murine and human cells, this long non-coding RNA is produced by pol I from the spacer promoter (SPr) located around 2 kb upstream of the transcription start site (TSS) [4] (shown in Fig. 1B, C). The coding region of pRNA starts at SPr and ends downstream of TSS. The primary 2,000 nt transcript is processed and reduced into a 150-250 nt mature pRNA (mpRNA) complementary to the region, which includes proximal terminator T_0_ as well as 47 S rDNA promoter with its upstream control element (UCE) and core promoter element (CPE) (shown in Fig. 1D). The mpRNA is stabilized by binding to TIP5 (transcription termination factor I interacting protein 5), the large subunit of the nucleolar remodelling complex (NoRC), which mediates transcriptional silencing via DNA methylation and histone modification [4, 7–10]. Apparently, two interactions are involved in the 47S rDNA silencing by the mpRNA. On the one hand, assuming a specific hairpin structure, mpRNA binds to TIP5 somewhere between UCE and CPE; on the other hand, the same RNA molecule recognizes the complementary double-strand DNA at T_0_, with which it forms a triplex. Some authors still question the existence of such triplex, indicating, among other things, that T_0_ sequence with its irregular alternation of purines and pyrimidines is not favourable for the triplex formation [11]. Nevertheless, associations of pRNA with T_0_, which also binds transcription termination factor I (TTF-1) and with NoRC, seem to be necessary for the subsequent recruitment of DNA methyltransferases and other factors introducing the repressive marks at the rDNA promoter area. The DNA:RNA triplex may stabilize the mpRNA and facilitate its specific interactions with NoRC and TTF-1 [4, 8, 12]. Expression of pRNA seems to be variously regulated as well. For example, TTF1 may arrest transcription from the SPr by acting as a “roadblock“ [13].
Results of recent studies indicate that the sequence variability, which normally occurs in mammalian cells, may account for the selective transcription of different 47S rDNA units [14]. Certain single nucleotide variants (SNVs), e.g. at the position -413 close to the promoter, correlate with the epigenetic marks and thus probably with the transcription activity [15–17]. Remarkably, very little attention has been paid so far to the upstream part of pRNA gene which does not belong to the mpRNA coding sequence. But this part, located between SPr and T_0_, contains a number of functional and potentially functional regions [14], including a whole micro-RNA gene, two sequences aligned with the exogenous micro-RNAs, two regions (Q1 and Q2) of short tandem repeats (CCCT)n (shown in Fig. 1), apparently prone to quadruplex formation. In our earlier work, we found that deletions in Q1 and Q2, as well as certain SNVs in the pRNA gene, significantly correlate with DNA methylation, suggesting that these variants may be engaged in the regulation of rDNA transcription. In the present study, by comparing the sequence of a segment of the pRNA gene with the sequence of RNA produced from it, we established directly the effect of DNA variability on the expression of pRNA and thus (indirectly) on the production of ribosomal RNA. Our new data also compelled us to revise our views on the role of DNA methylation in the economy of the rDNA locus, and we accordingly corrected the hypothesis formulated in our previous work [14].
Methods
Cell culture
The human fibrosarcoma cell line, HT1080, was obtained from the American Type Culture Collection (Rockville, MD, USA). The human primary fibroblast cell line, WI38, was obtained from Merck (Darmstadt, Germany). The cells were maintained in complete DMEM Dulbecco’s modified Eagle medium containing 10% FBS and 1% penicillin-streptomycin. All the chemicals were obtained from Merck Life Science (Prague, Czech Republic) unless otherwise stated. Cell cultures were maintained at 37 °C in a humidified atmosphere of 5% CO_2_.
Extraction of nucleic acids
Total DNA and RNA were sequentially extracted from the cells by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. Nucleic acids were quantified using the Eppendorf Biophotometer. Purity was considered sufficient if A260/A280 measured between 1.8 and 1.9 in DNA fractions and 1.9-2.0 in RNA fractions. RNA integration was additionally checked by gel electrophoresis, while 2 bands of 18S and 28S represented satisfactory RNA quality. Then cDNA was synthesised using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA).
Amplification of regions Q1 and Q2 containing CCCT repeats
Both DNA and cDNA fractions were amplified by PCR with the primers listed in Table 1. PCR amplification was performed in 35 cycles, and the resulting amplicons were verified by agarose gel electrophoresis. The gels showed clean, strong bands at the expected positions, without visible degradation or nonspecific products.
Table 1. Primers used for PCR amplification and their position in reference to U13369.1PositionSize of the product (bp)Primers41,469–41,790321F′ CGTTCCCTGTGTT TCCTTCTR′ GCAGAATCGGTAGGCTCTTC41,748–42,033285F′ GTCTGTCTCTGC GTGGATTCR′ CGAAACCGTGAGTCGAGAAG
Bioinformatic analysis
The amplicons were subjected to library preparation by the sequencing provider (Novogene Co., Ltd., Munich, Germany) who also performed the quality control. Standard Illumina-compatible library preparation protocols were used, including end repair, adapter ligation, and indexing. Libraries were subsequently pooled and sequenced on an Illumina NovaSeq6000 platform, using 250PE protocol. The sequencing output was of high quality across all samples, with Q30 scores above 70% and low error rates (0.05–0.09%). The sequencing data were processed by a combination of well-established tools and an in-house Python script. The sequencing adapters were removed using Cutadapt [18]. This step also included quality- and length-filtering – reads that were shorter than 60 bases were discarded as well as the bases that had a quality lower than 18. Reads of satisfactory quality were mapped to the reference rDNA sequence U13369.1 using BWA-MEM [19]. All reads that did not cover the Q1 or Q2 regions were filtered out using SAMtools [20]. Then clustering was performed by CD-HIT-EST [21, 22]. As the input files for the Python script, we used the clustered reads that were mapped again and transformed using their CIGAR strings to show better their deleted regions. The size of the reads was reduced so they would contain only the Q1 or Q2 regions with 5 bp long flanking sequences. The reads were finally clustered again, but this time with the set of Python functions instead of CD-HIT-EST, which better suited our case. The output of this script was a text file containing representative sequences of the clusters along with the numbers of their occurrence in the original file and percentage of the occurrence. In each sample, the output text file also contained a reference sequence cut at the same position as the examined reads.
Results
- Short variants in the entire CCCT-repeat-containing amplicons and the corresponding cDNA in HT1080 cells
After isolation of DNA and RNA from the cells, we used two pairs of primers to amplify a segment of pRNA gene, the sequence situated between the positions of 41,469 and 42,033 (referring to the human rDNA GenBank sequence U13369.1: https://www.ncbi.nlm.nih.gov/nuccore/U13369.1?report=graph&from=19514) containing two regions (Q1 and Q2) of CCCT repeats. The PCR products were subjected to deep sequencing to establish all variants and compare their frequencies in the DNA and the cDNA derived from the isolated RNA.
Examination of single nucleotide variants (SNVs) as well as short insertions and deletions (indels) in the regions of interest (shown in Fig. 1), revealed only one significant variant, C>T substitution at the position 41,574 (Fig. 1C; Table 2). This variant appeared with high frequency and reduced mononucleotide motif CCC to CCT. In our previous study [14] the same SNV_41,574_ showed a strong correlation with the DNA methylation status determined in the immunoprecipitation assay with antibody against 5-methylcytosine. In the present work we find that SNV_41,574_ occurs less frequently in the DNA than in the corresponding cDNA (Table 2).
Table 2. Occurrence of the C > T substitution (position 41,574) in DNA and cDNAFrequencyCoverageSignificanceDNA0.686,590,000p < 0.001cDNA0.951,830,000Meth0.481,195p* < 0.001Non-meth0.694,467 Depicts frequency of reads with high level methylation (meth) and low level methylation (non-meth) DNA according to the data of Chmurciakova et al. 2022
- (2)Variability of the CCCT repeat number in Q1 and Q2 regions and in the corresponding cDNA
To analyse the variability in the regions Q1 and Q2, we selected reads covering these microsatellite regions with at least 5 bp long flanking sequences. The observed variability was, for the most part, related to the number of CCCT repeats (shown in Table 3).
Table 3DNA and cDNA variants in the selected reads containing Q1 and Q2 regions with 5 bp long flanking sequencesThe CCCT repeats in the selected reads and in the reference sequence (Ref.) are marked by green and red respectively. Underlined are bases which differ from ref on account of SNVs and other variants in the flanking sequences
These data show that the number of CCCT repeats varied considerably more in DNA than in cDNA (shown in Table 4). Among the indels deletions predominated, thus most DNA reads had reduced numbers of the repeats. The inserts appeared only in Q1 and more frequently in DNA (8%) than in cDNA (2%) with p < 0.001.
Table 4. Variability of the CCCT repeat numbers in Q1 and Q2 regionsFrequencyQ1 DNAReference0.20Indels0.73Deletions0.65Insertions0.08Q1 cDNAReference0.95Indels0.04Deletions0.02Insertions0.02Q2 DNAReference0Indels0.98Deletions0.98Insertions0Q2 cDNAReference0Indels0.99Deletions0.99Insertions0The table shows the frequencies of the selected reads with repeat number corresponding to the reference sequence (Reference) and to indels. Only variants which appear in at least 2% reads are included
In Q2, all the reads contained reduced numbers of the repeats, which is probably the characteristic of the studied cell line [14]. But the length of indels differed in DNA and cDNA. To estimate this difference, we used the most frequent number of CCCT repeats (6) as a new reference (Ref#). We found that, in relation to Ref#, the indels appeared more frequently in DNA (7% deletions) than in cDNA (0% deletions) with p < 0.001 (Table 5). Thus, both Q1 and Q2 regions with reduced numbers of repeats were less likely to be expressed.
Table 5. Variability of the CCCT repeat number in the Q2 region with newly assumed reference (Ref#)FrequencyTotal DNA1DNA Ref#0.91DNA indels#0.07DNA deletions#0.07DNA inserts#0.01Total cDNA1cDNA Ref#0.97cDNA indels#0.01cDNA deletions#0cDNA inserts#0.01
- (3)Variability in the flanking sequences
The flanking sequences (Fls) of the two CCCT repeat regions are very similar. In fact, most of the 10 upstream nucleotides and 6 downstream nucleotides related to Q1 and Q2 respectively are identical (underlined):
Q1: (5‘)TTTTCTTCCT (CCCT)n GCTCCC(3‘).
Q2: (5‘)TGTTCTTTCT (CCCT)n GCTTCC(3‘).
But upstream of the 10 marked base pairs and downstream of the 6 marked base pairs, the similarity between Q1 and Q2 abruptly vanishes. It seems that each microsatellite (CCCT)n, together with its 16 adjacent base pairs, constitutes one functional or potentially functional unit. Assembling the reads containing at least 10 nt upstream and 6 nt downstream Fls of the repeats, we found the frequencies of the Fls‘ variants (Table 6).
Table 6. Variability of the flanking sequences (including 10 nt upstream and 6 nt downstream) in the Q1 and Q2 regions. Relative numbers of the reads corresponding to the reference sequence (Reference) and differing from it (Variants)Q1 DNAQ1 cDNAReference0.790.97Variants0.150.02Q2 DNAQ2 cDNAReference1.001.00Variants0.020.00
Only 3‘ flanking sequences contained significant variants, most of which occurred at the position 41,878. In Q1 region, Fls in DNA were more variable (15%) than Fls in cDNA (2%) with p < 0.05. In Q2, SNVs appeared rarely and only in DNA. Thus DNA proved to be more variable than the corresponding cDNA in Fls as well as in the (CCCT)n regions.
- (4)Variability of the entire pRNA locus in the WI38 cells
To examine whether the sequence variants identified in this work were specific for the tumour-derived cells (HT1080) or represented a more common phenomenon, we examined the variability in WI38 cells, a normal diploid cell line derived from human fibroblasts. The results were also compared with the findings of our earlier work [14], in which we examined correlation of sequence variability with DNA methylation in the entire pRNA locus situated approximately between the position -2 kb and the transcription start site. Sequencing of the pRNA locus in WI38 revealed totally 23 single nucleotide substitutions and indels (against 32 for the same region in HT1080 cells), 13 of these variants appeared with frequency close to 100%. Among the latter, 8 were found also in the HT1080 cells (Table 7), including insert 41,006 and SNV_41,008_ belonging to 3’ end of Alu element. Remarkably, two SNVs common two both cell lines, 41,574 and 42,329, correlated with DNA methylation in HT1080 cells.
Table 7. Variants detected in the pRNA locus of WI38 cellsPositionVariantFrequencyCoverage 41,006
insG
1.00
299
41,008
G > C
1.00
299
41,208
C > T
0.62
627
41,214
T > C
0.42
615 41,273T > G0.2554141,294T > G0.71520 41,310
delC
1.00
431
41,560
delT
1.00
244
41,574*
C > T
0.31
236
41,594
delT
0.97
233 41,659T > G0.25204 41,663
insTCCTC
0.56
126
41,665
delTCCCTCC
0.10
174
42,255
C > G
1.00
679
42,256
G > C
1.00
674
42,329*
C > A
0.10
288 42,398insC0.98521 42,400
C > G
0.98
505 42,418insGCG0.9953442,451insG0.9956442,455insCG0.9955942,500delA0.9956242,553delGCGGTC0.34435Variants in bold were also discovered in HT1080. Asterisks indicate SNVs which correlated with DNA methylation in HT1080 (see Table 3)
Other variants, such as 42,329 fall near the putative miRNA binding site (hsa-miR-6724) (Table 6). Moreover, the number of CCCT repeats in the Q1 region proved to be highly variable owing to the insertions and deletions at positions 41,663 and 41,665 respectively. In the Q2 region, large deletions affecting tetrameric CCCT motifs were also observed. However, due to low coverage in this part of the URR in WI38, these events could not be confidently characterized and are not included in the table.
Thus we found that pRNA locus in the normal WI38 cells was less variable than in the transformed HT1080 cells, but > 50% single nucleotide substitutions and indels constituted in WI38 were shared with HT1080. Importantly, some of these shared variants (SNV_41,574_, SNV_42,329_, indels in the Q1 region) appeared in the putatively functional sequences.
Discussion
The presented results indicate that deletions in (CCCT)n regions and other variants within the human pRNA gene inhibit its expression. This may be occasioned by the effects of the observed variations on the pRNA transcription, as well as on its processing, for example, through altering the cleavage sites. Then our data would really reflect a reduction of expression for certain intermediary (or intermediaries) of pRNA produced from the DNA with deletions. Nevertheless, such reduction would also signify the diminishing level of the final product, the mature pRNA.
The results presented in the Tables 2, 3, 4, 5, 6 and 7 shed a new light on the data of our earlier study where we found a significant correlation between the variants in the pRNA encoding sequence and DNA methylation [14]. On the one hand, we can reaffirm our previous conclusion that sequence variability in the pRNA encoding region can alter the expression of the known inhibitor of ribosomal genes. For we show directly that the variability of the region situated upstream of the rDNA promoter has a significant impact on the expression of pRNA. According to our data, the SNV_41,574_, which converts CCC motif into CCT (Fig. 1; Table 2) and deletions which reduce the numbers of (CCCT) repeats in the Q1 and Q2 regions (Fig. 1; Tables 3, 4 and 5), being very frequent in the DNA, occur but rarely in the respective transcripts. Thus, both SNV_41,574_ and (CCCT) deletions inhibit pRNA expression, and apparently promote rDNA transcription.
On the other hand, we originally believed that DNA methylation in the entire Upstream Regulatory Region (URR) extended between the position -2 kb and the transcription start site, should inhibit transcription of ribosomal genes [14]. But, there was no strong evidence for that. Only certain methylated CpG islands within the rDNA promoter (e.g., at the position -133 in mouse cells) have been experimentally identified as specific targets of the regulatory methyltransferases [23]. The data of the present study indicate that the variants located within the URR (Fig. 1C, D), which negatively correlated with DNA methylation (Table 2), also positively correlate with the transcription of pRNA and, therefore, must facilitate the 47S rDNA transcription. In our earlier work [14], we also suggested that the G-quadruplexes in the intact Q1 and Q2 regions should inhibit production of the pRNA transcript, and that various deletions of the CCCT repeats should destabilize the quadruplexes and promote pRNA expression. Now we find that such deletions, on the contrary, considerably diminish expression of pRNA (Tables 4 and 5).
To make sense of our earlier and present results, we will consider the following facts. Firstly, it is known that the length of short tandem repeats in various transcription regulating regions is a significant factor in the promoter activity [24–26]. Secondly, it was found in experiments with push-apart constructs that changing the distance between the TSS of the 47S rDNA region and the proximal terminator T_0_ by as little as 4 nucleotides can switch the rDNA promoter between active and silent states [27]. Thirdly, the (CCCT)n regions, which consist of the regular stretches of pyrimidine repeats, are optimal for the formation of parallel and antiparallel RNA-DNA:DNA triplexes. Such structures composed from the (CCCT)n /(GGGA)n regions of DNA and the (GGGA)n sequences of pre-pRNA, like the triplex formed between T_0_ and the complementary sequence of the mature pRNA, may transiently anchor the primary transcript to facilitate its processing and to prevent its drifting away to other rDNA units. Accordingly, we suppose that the tetrameric deletions in the Q1 and Q2 regions get suppressed, for they would damage the pRNA function by reducing the probability of the triplex formation. Signal to this suppression may be provided by shortening the distance, e.g., between SPr and T_0_. It must be mentioned that the pRNA transcription would hardly be prevented by the construction of triplex, since its RNA component is rather short-lived: according to our earlier study, the half-life time of various parts of pre-pRNA is about 5 min [28].
Although the role of triplex formation based on Q1 and Q2 regions remains hypothetical, the data of this and our earlier publications, show that the transcription of pRNA, as well as rDNA, significantly depends on the variability of the pRNA encoding sequence situated 1–2 kb upstream of the rDNA promoter. Additionally, our data indicate that DNA methylation in these upstream loci can reduce the transcription of pRNA and thus promote the expression of rDNA. The presence of certain variations in the DNA sequence seems to be required for normal regulation of the rDNA synthesis.
Conclusions
Our study demonstrates that sequence variability of the human pRNA gene significantly impacts its expression. We show that specific variants, such as SNV41,574 and deletions in Q1 and Q2 regions of short tandem repeats (CCCT)n, are frequent in DNA but rarely detected in transcripts, suggesting selective inhibition of pRNA expression and, consequently, promotion of rDNA activity. Furthermore, our findings revise our previous supposition about the role of G-quadruplex structures and DNA methylation at the loci situated upstream of the promoter. Thus our new findings indicate that methylation at the position 41,574 should promote the 47S rDNA transcription. This work emphasizes the regulatory importance of the pRNA encoding region, located 1–2 kb upstream of the rDNA transcription start site.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bersaglieri C, Santoro R. Genome organization in and around the nucleolus. Cells. 2019;8(6).10.3390/cells 8060579 PMC 662810831212844 · doi ↗ · pubmed ↗
- 2Sibai DS, Tremblay MG, Lessard F, Tav C, Sabourin-Felix M, Robinson M et al. TTF 1 control of Lnc RNA synthesis delineates a tumor suppressor pathway directly regulating the ribosomal RNA genes. J Cell Physiol. 2024.10.1002/jcp.3130338764354 · doi ↗ · pubmed ↗
- 3Chmurciakova N, Smirnov E, Kurfurst J, Liska F, Cmarko D. Variability of human r DNA and transcription activity of the ribosomal genes. Int J Mol Sci. 2022;23(23).10.3390/ijms 232315195 PMC 974079636499515 · doi ↗ · pubmed ↗
- 4Rodriguez-Algarra F, Seaborne RAE, Danson AF, Yildizoglu S, Yoshikawa H, Law PP et al. Genetic variation at mouse and human ribosomal DNA influences associated epigenetic States. Genome Biol. 2022;23(1).10.1186/s 13059-022-02617-x PMC 884254035164830 · doi ↗ · pubmed ↗
- 5Parks MM, Kurylo CM, Dass RA, Bojmar L, Lyden D, Vincent CT et al. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci Adv. 2018;4(2).10.1126/sciadv.aao 0665 PMC 582997329503865 · doi ↗ · pubmed ↗