An Orally Administered Misuse Deterrent Opioid Prodrug for Treatment of Acute Pain
Douglas A. Rose, Joseph W. Treacy, Karina Seth, Anthony F. Tanzillo, Allison Li, Kyle Tamshen, Lily K. Sloan, Natalie Boehnke, Christopher J. Evans, Catherine M. Cahill, Heather D. Maynard

TL;DR
This paper introduces a new opioid prodrug designed to prevent misuse by requiring specific conditions for activation, offering a safer option for treating acute pain.
Contribution
The development of a novel oxycodone prodrug with three covalent misuse deterrent layers that resist common subversion methods.
Findings
The prodrug is resistant to degradation by acidic and basic pH, household chemicals, and enzyme supplements.
The prodrug shows analgesic effects in mice only after oral administration, not intraperitoneal injection.
The dual-enzyme requirement for activation suggests strong misuse deterrent properties.
Abstract
Opioids are a powerful class of medicines due to their ability to alleviate acute pain. However, the use of prescription opioids has led to an epidemic in the United States. Many efforts to combat this are ongoing, including the preparation of misuse deterrent opioid formulations, some of which can be subverted using common household chemicals. Herein, the development of an oxycodone-containing peptide prodrug is reported that contains three covalent misuse deterrent protective layers. This prodrug is resistant to degradation in the presence of acidic and basic pH conditions, common household chemicals, and enzyme supplements. After optimization of the peptide sequence, the lead prodrug is composed of a branched lysine residue coupled to a tert-butyl-protected tyrosine that is not naturally recognized by digestive enzymes. There is a necessary sequence of protecting group removal and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1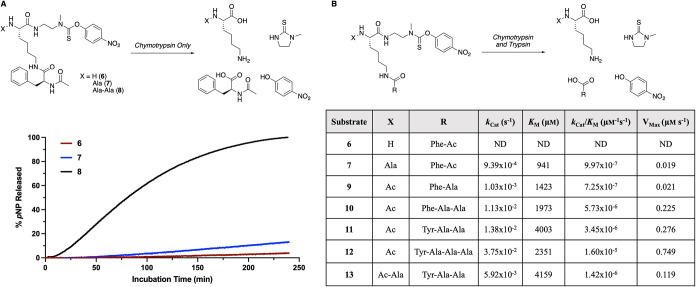 2
2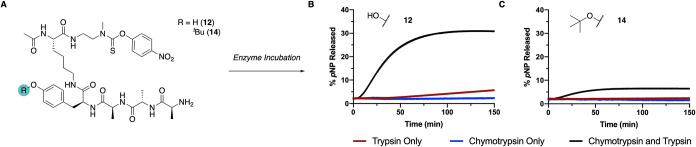 3
3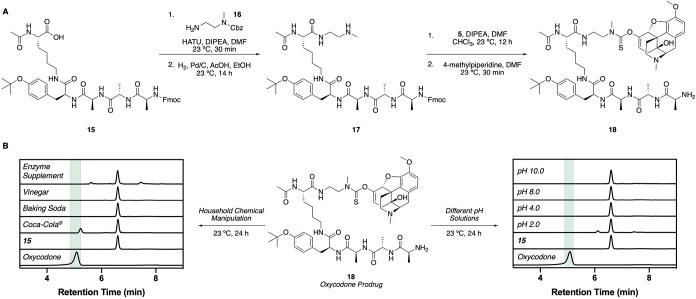 4
4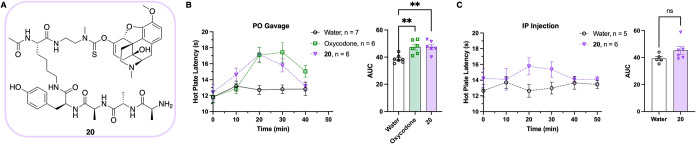 5
5- —National Institutes of Health10.13039/100000002
- —National Institutes of Health10.13039/100000002
- —Division of Chemistry10.13039/100000165
- —California NanoSystems Institute10.13039/100018432
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPain Management and Opioid Use · Opioid Use Disorder Treatment · Pain Mechanisms and Treatments
Introduction
The United States has a tumultuous past with opioids dating back to the late 19th and early 20th centuries after the first chemical isolation of heroin from opium in 1898.? The consumption and production of semisynthetic and synthetic opioids have evolved over time, but they still remain highly addictive substances. In 2021, the CDC reported more than 16,500 prescription opioid-associated overdose deaths in the United States, and in 2023, 8.9 million people were reported to have misused opioids8.2 million of whom misused prescription opioids. ?,? The increase in the prescription opioid use in the United States over the past few decades has been traced back to multiple factors, ?−? ? and the use of prescription opioids can initiate the transition of users to injection-based opioids such as heroin and fentanyl. ?−? ? ? ? ? In 2017, the US Department of Health and Human Services formally recognized the opioid epidemic as a national health crisis. Consequently, a plan of action was implemented to reverse the damage done by this class of pharmaceuticals.? As a result, there has been an increase in efforts to develop new drugs or drug delivery systems to combat this crisis. Specifically, one area of research is the study of nonopioid-based treatments that target proteins involved in nociception or inflammation signaling pathways. ?,? Recently, the FDA approved the highly selective Na_V_1.8 pain signal inhibitor JOURNAVX (suzetrigine) for the treatment of acute pain.? Another alternative is the development of opioid formulations that contain misuse deterrent engineering controls, which are designed to mitigate the typical forms of misuse by increasing the difficulty of extracting large amounts of active opioid for instant release in pursuit of the euphoric effects. ?−? ? ?
The FDA has described five general strategies for developing misuse deterrent formulations including physical or chemical barriers to mechanical alteration, agonist/antagonist combinations, coformulation with aversive substances released upon tampering, unconventional delivery systems (e.g., subcutaneous implants), and prodrugs that are only activated following oral administration.? There are currently four formulations with FDA-approved labeling describing misuse deterrent properties, three with oxycodone and one with hydrocodone as the opioid. ?,? All of these formulations rely on a noncovalent encapsulation strategy, wherein the opioid is sequestered within a porous polymeric network and slowly released to prevent the rapid onset of euphoria while still maintaining the analgesic properties of the opioid. However, many of these engineering controls can be circumvented by knowledgeable users employing household supplies, thereby reducing their effectiveness. ?,?
A promising alternative to these noncovalent misuse deterrent strategies is to employ covalent modifications in the form of a prodrug. Prodrug strategies have been widely used in cancer drug delivery and antimicrobials, ?−? ? ? ? ? whereas fewer examples have been developed in opioid delivery and prevention of opioid overdose. ?−? ? ? However, these prodrugs could provide targeted and controlled release of the opioid to prevent a burst release that may lead to an adverse health event. Designing the prodrug to target opioid release within the gastrointestinal tract may safeguard against intravenous and intranasal routes of misuse. This covalent prodrug strategy is currently being pursued by two separate companies, all of which are in phase three clinical trials.? Two of these prodrugs take advantage of the acidic conditions in the stomach to cleave either an ester or phosphoester modification of the opioid. However, these strategies can be circumvented through chemical manipulations using household supplies, allowing users to bypass the engineering controls. The third prodrug relies on trypsin, a protease found in the small intestine, to enzymatically cleave the prodrug and release oxycodone. ?,? This prodrug has an enhanced misuse deterrence profile,? but it still relies on one stimulus to release the active opioid.
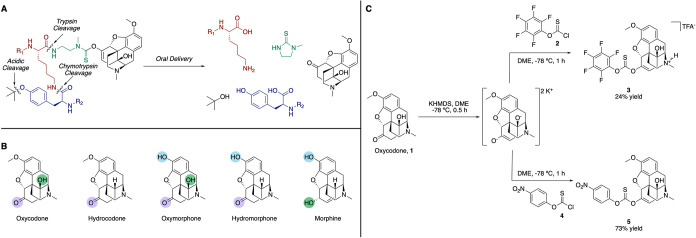
To address these shortcomings and add further layers of protection,? we have designed a multiple stimuli-responsive, misuse deterrent opioid prodrug (FigureA). This strategy relies upon a covalent linkage of oxycodone to an enzymatically labile peptide, whereupon sequential incubation with acid and the digestive enzymes trypsin and chymotrypsin, the native opioid is released. Critically, this strategy is only amenable to oral delivery of the prodrug, and it should not provide analgesic effects when delivered intravenously, thus discouraging users from misusing the opioid. We envisioned that the acidic conditions in the stomach would remove the tert-butyl ether group from tyrosine, allowing chymotrypsin to recognize and cleave the isopeptide bond between the tyrosine residue and the ε-amine of a lysine residue.? Once cleaved, the lysine residue can further be digested by trypsin, which cleaves the lysine C-terminally and releases a primary amine. ?,? This primary amine can then rapidly cyclize ?,? and release the active opioid, forming 1-methylimidazolidine-2-thione as a nontoxic byproduct.?
(A) Proposed scheme of misuse deterrent opioid prodrug design relying on three protective layers. (B) Evaluation of functional groups amenable to reversible derivatization among five of the most commonly prescribed opioids. (C) Synthesis of oxycodone electrophiles 3 and 5 for coupling to the peptide prodrug scaffold.
Results and Discussion
Evaluation of Common Opioids for Reversible Modification
Examining the structures of five of the most commonly prescribed opioids reveals multiple points for derivatization and subsequent attachment to the peptide scaffold.? Conserved across four of these opioids is the ketone motif, which we identified as a potential reversible linkage point for the peptide (FigureB). By forming the enolate, we could generate a nucleophilic moiety and couple it to a thionochloroformate or chloroformate electrophile. This electrophile could then react with an amine to form a stable thionocarbamate or carbamate linkage and the covalent prodrug. With oxycodone being the most commonly used opioid in the misuse deterrent formulations with FDA-approved labeling, we attempted a selective modification of the ketone of oxycodone (1). An initial investigation showed that the tertiary alcohol of oxycodone was unreactive toward modifications, enhancing our optimism that the enolate strategy would be ideally suited for selective modification of oxycodone. We also observed that the thionocarbonate product was less susceptible to hydrolysis compared to the carbonate (data not shown), likely due to its lower electrophilicity, so the thionocarbonate was chosen for the synthesis of all oxycodone-containing electrophiles going forward.
To ensure selective modification of the O-enolate, we screened conditions from previous reports, which indicate that the use of a coordinating solvent and a hard electrophile favors modification of the O-enolate over the C-enolate.? We found that the use of potassium bis(trimethylsilyl)amide (KHMDS) as a base, dimethoxyethane (DME) as a solvent, and pentafluorophenyl O-thionochloroformate (2) as an electrophile minimized C-modification products and progressed to full conversion from the ketone. However, the thionocarbonate product 3 was isolated in only 24% yield (FigureC). The low yield following purification was likely due to the instability of 3 toward chromatography. Further optimization of the electrophile synthesis resulted in the use of p-nitrophenol O-thionochloroformate (4) that was amenable to purification via precipitation, which afforded activated oxycodone-containing thionocarbonate 5 in 73% yield (FigureC).
Optimization of the Peptide Sequence for Release
Using the branched lysine peptide sequences modified from our previous work,? we began to explore the sequence effects on the efficiency and release rate of a model reporter molecule, p-nitrophenol (pNP). Peptides 6–8 were initially prepared to determine whether the peptide backbone length had any effect on the release of pNP in the presence of chymotrypsin alone (FigureA; see Supporting Information (SI) for synthetic procedures). Chymotrypsin promiscuity (i.e., cleavage of both sites) could lead to less misuse deterrence as only one enzyme could promote the release of the opioid. Substrates 6 and 7 both showed less than 15% release over the course of 4 h (FiguresA, S13, and S14). However, substrate 8 reached greater than 90% pNP release within 3 h (FiguresA and S15). This illustrates that increasing the length of the peptide backbone by at least two amino acids directly facilitates chymotrypsin promiscuity, leading to the release of the pNP reporter. Whether the addition of these two alanine residues affected the conformation of the peptide sequence in the chymotrypsin binding pocket was not investigated; however, identifying a prodrug scaffold with minimal release in the presence of only chymotrypsin was successful (i.e., 6 and 7), so we proceeded toward further optimization of the prodrug sequence with this new structure–activity insight.
(A) Effect of the α-amine chain length on pNP release in the presence of chymotrypsin only. (B) Michaelis–Menten kinetics for various substrates in the presence of both chymotrypsin and trypsin. ND = not detectable. See SI for details on individual release assays.
In order to ensure the prodrug would be cleaved effectively once it was exposed to chymotrypsin and trypsin in concentrations found in the stomach,? we elected to optimize the peptide sequence further, particularly on the ε-amine of the lysine, to enhance the substrate efficiency (FigureB). This would help to ensure that oxycodone is released in the gastrointestinal tract. To examine substrate specificity, Michaelis–Menten kinetics for substrates 6 and 7 were determined in the presence of both chymotrypsin and trypsin, but there was negligible release over 4 h for 6, while 7 had a substrate specificity (k Cat/K M) of 9.97 × 10^–7^ μm ^–1^ s^–1^ (FigureB). Previous studies have shown that chymotrypsin binding can be enhanced through additional nonbulky amino acid groups following the aromatic residue. ?,?−? ? Accordingly, substrates 9 and 10, with alanine residues coupled to phenylalanine, were prepared, and their kinetics were examined. Both 7 and 9 showed comparable substrate specificity, but the additional alanine on 10 resulted in significantly enhanced specificity (5.73 × 10^–6^ μm ^–1^ s^–1^). Substitution of phenylalanine with tyrosine has been shown to increase substrate specificity with chymotrypsin, so we synthesized the tyrosine-containing substrate 11 and observed a slight decrease in substrate specificity relative to the phenylalanine analog 10. Coupling a third alanine to the tyrosine residue (12) increased the substrate specificity by around 5-fold compared to 11. Since coupling of alanine residues to the α-amine of 6–8 increased the percentage of pNP release (FigureA), 13 with a single alanine on the N-terminus was prepared. Compared to substrate 11 with no N-terminal alanine, there was a minimal effect on the K M, but the V Max was reduced by half, leading to a lower k Cat/K M. Since 12 had the highest k Cat/K M and the fastest V Max, we proceeded to use this peptide scaffold for further experiments.
Addition of a Third Protective Layer for Misuse Deterrence
While the addition of another alanine residue on the ε-peptide chain of 10 may result in a substrate specificity comparable to that of 12, the use of tyrosine incorporated a phenol into the peptide, which could be functionalized to add an additional layer of misuse deterrence within the prodrug. We hypothesized that by protecting the phenol with a tert-butyl ether, chymotrypsin would not recognize the tyrosine derivative prior to passage through the acidic conditions in the stomach where the tert-butyl ether would be removed.? This additional security measure removes the ability to pretreat the prodrug with store-bought digestive enzyme supplements to release the opioid. To probe this hypothesis, in vitro simulated digestion assays were carried out with 12 (−OH) and 14 (−O^ t ^Bu) in the presence of trypsin, chymotrypsin, or both enzymes (FigureA). pNP release from 12 was only observed in the presence of both proteolytic enzymes (FigureB), validating the necessity of both chymotrypsin and trypsin for the release of pNP. However, the presence of a tert-butyl ether in 14 protecting the phenol greatly reduced the amount of pNP released (∼5% release over 150 min), confirming that an additional acidic pretreatment is required for this prodrug scaffold to release the model reporter (FigureC).
(A) Enzymatic release assay of pNP-containing peptides 12 and 14. (B) pNP release curves from the 12. (C) pNP release curves from 14.
Misuse Deterrence Testing of Prodrugs against Common Chemicals
To determine if this peptide scaffold was stable to different pH solutions and household chemicals that can break down other misuse deterrent formulations, the oxycodone-containing prodrug was synthesized (FigureA; see SI for details). Peptide 15 was synthesized by Fmoc solid-phase peptide synthesis using a 2-chlorotrityl chloride resin. Notably, the N-Fmoc group from the final coupling was left intact, and the O^ t ^Bu group was also retained through the selective cleavage of the peptide from the resin using hexafluoroisopropanol. Coupling of 16 and subsequent chemoselective deprotection of the Cbz group afforded 17. The amine then underwent selective formation of the thionocarbamate using the oxycodone-containing electrophile 5, and then deprotection of the Fmoc group generated the oxycodone-containing prodrug 18 with the O^ t ^Bu group intact in four linear steps from peptide 15 (FigureA). To examine the stability of this construct, 18 was incubated for 24 h at 23 °C with a variety of household chemicals and showed no detectable release of oxycodone (FigureB; see SI for details). Additionally, a store-bought digestive enzyme kit was unable to cleave the prodrug and to provide any release of oxycodone (FigureB). Prodrug 18 was also stable in different buffers varying from pH 2.0 to 10.0 with no observable release of oxycodone over 24 h at 23 °C (FigureB). The misuse deterrence profile of this prodrug indicates that it is challenging to extract oxycodone from this prodrug scaffold using commonly available chemicals.
(A) Synthetic scheme of prodrug 18 from peptide 15. Salts of the isolated amines are not shown for the sake of clarity. See SI for further details on the synthesis of 18. (B) Household chemical and pH-based manipulation of oxycodone-containing prodrug 18. HPLC traces are shown at 254 nm. The peak at 5.3 min in the Coca-Cola run corresponds to aspartame.
Peptide Byproduct Inhibition Studies
Prior to moving forward with analgesia studies in vivo, it was important to ensure that the AAAY (19) peptide byproduct from chymotrypsin cleavage was not competitively inhibiting the pNP release. This could be an issue in vivo, as the PEPT1 transepithelial transporter in the digestive tract has a very weak binding affinity for tetrapeptides.? A lack of transport for 19 could lead to the accumulation and generation of a competitive inhibitor for chymotrypsin, thus reducing the overall amount of opioid released. To examine this hypothesis, Michaelis–Menten competitive inhibition enzyme kinetics were performed with chymotrypsin and trypsin for 12 in the presence of peptide 19. The activity of 12 proved to be negligibly affected by the presence of 19 (see SI, Figure S30). These observations enhanced our confidence in using the 12 prodrug scaffold for in vivo analgesia studies.
In Vivo Determination of Analgesic Effects
The use of 18 for in vivo studies was precluded due to the decreased acidity of the mouse stomach compared to the human stomach (ca. pH 4.0 compared to 1.5 of the empty human stomach).? This less acidic environment does not cleave the tert-butyl ether that is necessary for the enzymatic recognition of tyrosine, and thus prevents the release of oxycodone. Accordingly, the synthesis of prodrug scaffold 18 was modified to remove the tert-butyl group and afford the oxycodone prodrug 20 (FigureA; see SI for details). Hot plate latency, a measure of acute pain response, was used to determine the antinociceptive effects of the oxycodone prodrug.? Oxycodone was administered as a positive control at a dose of 3 mg/kg (PO gavage). Since the in vitro simulated digestion assays stagnated at ∼30% release of pNP from 12 (FigureB), a higher dose of 30 mg/kg of oxycodone in 20 was used to determine the analgesic effects of the prodrug. Results for 20 indicate that the thermal threshold latency was increased to a level similar to that of the oxycodone positive control. This indicates that the oxycodone is released in vivo and is effective in inducing an analgesic effect in mice that is statistically significant from the PO gavage containing only water (FigureB). The analgesic time course for 20 is similar to that of oxycodone, which indicates that it can be effective in the rapid treatment of acute pain (FigureB).
(A) Structure of oxycodone-containing prodrug 20 used for in vivo analgesia experiments. (B) Hot plate latency data and statistical analysis for PO gavage of 20 performed at 30 mg/kg of oxycodone in prodrug and 3 mg/kg of oxycodone. ** = p < 0.01. (C) Hot plate latency data and statistical analysis for IP injection of 20 performed at 30 mg/kg of oxycodone in prodrug. ns = not significant.
To confirm that this analgesic effect was due to enzymatic degradation of the prodrug in the digestive system, the antinociceptive effects of prodrug 20 were examined following systemic intraperitoneal (IP) injection. This route of administration produced withdrawal threshold latencies similar to those of the water control despite a moderate initial increase in the hot plate latency from 20 (FigureC). In combination with previous reports demonstrating that there is an analgesic effect with the IP injection of oxycodone, ?,? these results indicate that the oxycodone release is due to enzymatic cleavage of the prodrug in the stomach, corroborating the specificity for oral administration of the prodrug.
Conclusions
We have developed an orally administered dual-enzyme-responsive peptide–oxycodone prodrug. The design of this prodrug relies on an initial passage through the acidic conditions in the stomach to activate the peptide sequence, followed by the enzymatically triggered release of free oxycodone. Improvement in the prodrug design was carried out through peptide sequence optimization to ensure the requirement of both chymotrypsin and trypsin while simultaneously enhancing the payload release kinetics. Through the preparation of a pNP oxycodone O-thionochloroformate electrophile (5), the ketone in oxycodone was functionalized to generate a reversible oxycodone modification that could be incorporated into a peptide scaffold. Optimized prodrug 20 was evaluated for its analgesic properties in vivo where it demonstrated that there was a similar effect to that of oxycodone upon oral administration of prodrug 20. However, 20 did not alleviate acute pain upon IP injection. As misuse of oxycodone is often performed via intravenous injection, this feature of the prodrug contributes to its misuse deterrent profile. Additionally, this prodrug was stable to a broad pH range, household chemicals, and a digestive enzyme kit, demonstrating that oxycodone was not readily extracted. This observation meaningfully enhances its ability to act as a misuse deterrent prodrug.
Further development of the peptide scaffold to improve release fidelity, while retaining the dual-enzyme specificity, may allow for this prodrug to be used in vivo at a comparable dosage to oxycodone. Additional release assays in vitro could provide a more comprehensive misuse deterrence profile for the prodrug scaffold outlined herein. Further testing of 20 in postoperative or chronic pain models merits further examination to determine the potential for this prodrug to treat different types of pain. We propose that this general strategy of ketone modification for electrophile synthesis should also be amenable to other opioids and small molecule therapeutics. The fundamental chemistry and misuse deterrent opioid prodrug developed herein are anticipated to complement and advance existing efforts to combat the ongoing opioid epidemic.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Heroin Crisis (1898–1922) . Topics in Chronicling America. https://guides.loc.gov/chronicling-america-heroin-crisis#:~:text=Drugs%20derived%20from%20opium%20are,spreading%20rapidly%20with%20tragic%20consequences.
- 2National Institute on Drug Abuse . Drug Overdose Death Rates, 2023. https://nida.nih.gov/research-topics/trends-statistics/overdose-death-rates.
- 3Substance Abuse and Mental Health Services Administration . Results from the 2023 National Survey on Drug Use and Health (NSDUH): Key Substance Use and Mental Health Indicators in the United States, 2024. https://library.samhsa.gov/product/2023-nsduh-report/pep 24-07-021.
- 4Van Zee A.The Promotion and Marketing of Oxy Contin: Commercial Triumph, Public Health Tragedy Am. J. Public Health 200999222122710.2105/AJPH.2007.13171418799767 PMC 2622774 · doi ↗ · pubmed ↗
- 5De Weerdt S.Tracing the US Opioid Crisis to Its Roots Nature 20195737773 S 10S 1210.1038/d 41586-019-02686-231511672 · doi ↗ · pubmed ↗
- 6Kibaly C.Alderete J. A.Liu S. H.Nasef H. S.Law P.-Y.Evans C. J.Cahill C. M.Oxycodone in the Opioid Epidemic: High ‘Liking’, ‘Wanting’, and Abuse Liability Cell. Mol. Neurobiol.202141589992610.1007/s 10571-020-01013-y 33245509 PMC 8155122 · doi ↗ · pubmed ↗
- 7Cicero T. J.Ellis M. S.Surratt H. L.Kurtz S. P.The Changing Face of Heroin Use in the United States: A Retrospective Analysis of the Past 50 Years JAMA Psychiatry 201471782182610.1001/jamapsychiatry.2014.36624871348 · doi ↗ · pubmed ↗
- 8Banerjee G.Edelman E. J.Barry D. T.Becker W. C.CerdáM.Crystal S.Gaither J. R.Gordon A. J.Gordon K. S.Kerns R. D.Martins S. S.Fiellin D. A.Marshall B. D. L.Non-Medical Use of Prescription Opioids Is Associated with Heroin Initiation among US Veterans: A Prospective Cohort Study: Non-Medical Opioid Use and Heroin Initiation Addiction 2016111112021203110.1111/add.1349127552496 PMC 5056813 · doi ↗ · pubmed ↗