Molecular Characterization of Plasmodium Species to Strengthen Malaria Surveillance in Migrant Populations in Honduras
Ashley Godoy, Kevin Euceda, Alejandra Pinto, Hugo O. Valdivia, Lesly Chaver, Gloria Ardon, Gustavo Fontecha

TL;DR
This study uses molecular markers to distinguish local and imported malaria cases in Honduras, aiding surveillance in migrant populations.
Contribution
The study identifies novel genetic markers in Plasmodium species that help differentiate local from imported malaria infections.
Findings
Polymorphisms in P. falciparum markers like pfmdr1 and pfs47 distinguish migrant from local infections.
Novel P. vivax haplotypes, including VK210 and VK247 variants, were detected in migrant samples.
Markers such as pvs47 and pvs48/45 proved useful for inferring geographic origin of infections.
Abstract
As Honduras approaches malaria elimination, imported infections pose a growing challenge to disease surveillance and control. In this study, we analyzed 14 molecular markers—six from Plasmodium falciparum and eight from P. vivax—in samples from local and migrant subjects to assess their utility in differentiating local versus imported infections. All P. falciparum isolates carried the wild-type pfcrt haplotype associated with chloroquine susceptibility. However, polymorphisms in pfmdr1, pfama1, pfglurp, and pfs47 revealed distinct genotypes in migrant versus local samples, suggesting external origins. For P. vivax, three novel pvcsp VK210 haplotypes and the first detection of a VK247 variant in Honduras were identified in migrants. Additional novel haplotypes were found in pvmsp1, pvmsp3α, pvmsp3β, pvs47, and pvs48/45. Several of these markers—particularly pfmdr1, pfs47, pvs47, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
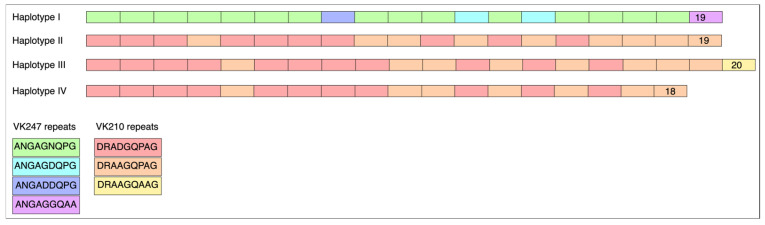 Figure 1
Figure 1- —the Genetic Research Center
- —CIG-UNAH
- —the Armed Forces Health Surveillance Division (AFHSD)
- —Global Emerging Infections Surveillance (GEIS) Branch
- —DICIHT-UNAH
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMalaria Research and Control · Mosquito-borne diseases and control · Vector-borne infectious diseases
1. Introduction
Malaria remains a significant public health challenge in the Americas, although the region has achieved notable reductions in transmission over the past two decades [1]. Honduras is one of the countries in Central America where malaria continues to be endemic, primarily caused by Plasmodium vivax, with less than 10% of cases of P. falciparum. In recent years, the country has experienced a marked decline in malaria incidence, accompanied by a narrowing of the geographic range and a reduction in parasite genetic diversity [2,3]. These changes have important implications for malaria control and elimination strategies, including surveillance, diagnostics, and treatment policies.
At the same time, increasing human mobility, particularly among individuals transiting through Honduras toward North America, poses new challenges for malaria surveillance [4,5]. Individuals traveling from high-transmission areas in Africa, Asia, and South America may carry genetically distinct parasite strains into regions with limited transmission, such as Honduras [6]. The introduction of non-native Plasmodium strains has potential consequences for drug resistance, diagnostic effectiveness, and transmission dynamics. Thus, there is a need to distinguish between locally acquired infections and those imported from other regions, especially in the context of elimination efforts [7].
Conventional epidemiological surveillance in Honduras is led by the Ministry of Health through standardized protocols, where thick blood smear microscopy remains the gold standard for malaria diagnosis and case confirmation at the national level [8]. National surveillance manuals and operational guidelines emphasize case detection, treatment, and reporting through the health information system. While microscopy is reliable and cost-effective, it has limitations in detecting low-density and mixed infections. In this context, molecular approaches can complement conventional surveillance by improving detection sensitivity and providing species-level identification, which is particularly relevant in migrant populations that may introduce Plasmodium species not commonly found in Honduras.
Molecular tools have become essential in characterizing malaria parasite populations and tracking transmission dynamics. Several genetic markers, including drug resistance genes [9,10], surface antigens [3,11,12], and vaccine candidates [13,14,15,16], have been used to infer parasite origin, assess population structure, and detect novel variants. In settings with limited access to next-generation sequencing (NGS) technologies [17], targeted sequencing of informative loci offers a cost-effective alternative for genetic surveillance. In Honduras, these molecular markers have been employed for over two decades [2,3,15,18,19,20,21,22,23,24,25,26], providing a valuable baseline for comparative genetic analyses.
Therefore, the objective of this study was to evaluate the utility of 14 molecular markers—six for P. falciparum and eight for P. vivax—to distinguish between local and potentially imported malaria infections. We analyzed samples collected from both local patients and migrant individuals transiting through Honduras. By comparing these sequences to existing national and international databases, we sought to determine the informativeness of each marker in inferring geographic origin. Additionally, the study contributes to building a reference framework of parasite genetic diversity within Honduras, which may support future efforts to differentiate between relapses and reinfections in patients treated with radical cure therapies.
2. Materials and Methods
2.1. Sample Collection
A total of 238 blood samples impregnated on Whatman No. 3 filter paper, previously diagnosed with malaria by the Honduran Ministry of Health, were analyzed. These samples, collected between 2023 and 2024, were evaluated as part of quality control for microscopy or rapid diagnostic test (RDT) malaria diagnosis using molecular techniques, as well as for the analysis of drug resistance-associated genes, in compliance with national regulations and routine disease surveillance [8]. According to national guidelines, malaria cases were defined as confirmed when parasites were detected by thick blood smear microscopy or RDT. Imported cases were defined as malaria infections diagnosed in individuals with a travel history to endemic countries outside Honduras during the 30 days preceding diagnosis. Both local and migrant patients were treated according to the national treatment scheme: chloroquine for three days plus primaquine for 14 days for P. vivax, and chloroquine for three days plus a single dose of primaquine for P. falciparum. However, treatment outcomes were not available, as this information was not systematically collected for the patients included in this study. Among the total samples, five were identified as originating from patients in the migration process through Honduras to North America; these five represent the entirety of imported malaria cases detected during the study period. The samples were anonymized, but limited demographic data were available for the patients. Without available NGS capabilities, these five samples were thoroughly characterized in this study using a panel of 14 molecular markers available in our laboratory. Also, four to seven local samples were analyzed using the same panel of molecular markers. This characterization aimed to assess the level of information each marker could provide to determine whether malaria was locally acquired or originated in another geographic region. The study was reviewed and approved by the ethics committee (CEI-MEIZ) of the National Autonomous University of Honduras (UNAH) under protocol number PI 12-2024.
2.2. DNA Extraction and Molecular Diagnosis
DNA extraction was performed using two 2 mm diameter circles that were punched from blood-impregnated filter paper. Genomic DNA was isolated using the Extracta^®^ DNA Prep for PCR kit (QuantaBio, Beverly, MA, USA) according to the manufacturer’s protocol and stored at −20 °C until analysis. Plasmodium genus detection was performed using PET-PCR with primers described in Table 1 [25,27]. Samples testing positive were further amplified with species-specific primers for P. falciparum and P. vivax, the two malaria parasites endemic to Honduras. PCR reactions were prepared in a 20 μL volume containing 10 μL GoTaq^®^ Probe qPCR Master Mix (Promega Corp., Madison, WI, USA), 0.5 μL each of forward and reverse primers (10 μM) (Table 1), 4 μL nuclease-free water, and 5 μL DNA template (~40 ng/μL). Amplification was carried out on a Mic qPCR Cycler (Bio Molecular Systems, Brisbane, Australia) under the following conditions. Initial denaturation: 95 °C for 15 min, 45 cycles of 95 °C for 20 s, 63 °C for 40 s, 72 °C for 30 s. Fluorescence detection used 6FAM-labeled primers for genus identification and HEX-labeled primers for species discrimination. All runs included positive and negative controls, with a cycle threshold (Ct) ≤ 42 defined as a positive result. Data were analyzed using Mic qPCR Cycler Software v2.10.1.3.
2.3. Molecular Markers for P. falciparum: pfcrt, pfmdr1, pfglurp, pfama1, pfhrp2/3, and pfs47
In total, one P. falciparum sample (M1) and four P. vivax samples (M2–M5) were analyzed using the molecular markers listed in Table 2 and Table 3, with the country of origin indicated for each sample.
One sample (designated “M1”) obtained from a foreign subject with falciparum malaria migrating through the country was analyzed. The subject was recruited in the Cortés department, though their country of origin and most recent geographic provenance remain unknown. Additionally, four samples from local patients were analyzed. A 264 bp region of pfcrt exon 2 (encoding amino acids 72–76), known to harbor polymorphisms linked to P. falciparum drug resistance, was amplified and sequenced using nested PCR (nPCR) with primers described in Table 1 [3,29]. Amplicons were visualized on 1% ethidium bromide-stained agarose gels, purified, and sequenced by Psomagen Inc. (Rockville, MD, USA) using primer AL5631 (Table 1). Sequences were analyzed in Geneious Prime v.2024.05 to assess residues 72–76 of pfcrt.
Three regions of the pfmdr1 gene were amplified to identify five polymorphisms at codons 86 and 184 (first segment), 1034 and 1046 (second segment), and 1246 (third segment). The three regions were amplified by nested PCRs according to a previous report [20] (Table 1). PCR products were resolved on 2% agarose gels stained with ethidium bromide and visualized under UV transillumination. The products were sequenced from both sides.
A semi-nested and a nested PCR were used to amplify a fragment of the P. falciparum glurp [3,11] and ama1 genes [3,37], respectively (Table 1). Amplicons were bidirectionally sequenced and subsequently aligned to generate a consensus sequence for each individual. The nucleotide sequences of pfglurp and pfama1 were translated into amino acid sequences using the correct open reading frame (ORF). The identified haplotypes were compared against those previously reported as circulating in the country. For any novel haplotypes discovered, sequences were deposited in GenBank, and accession numbers were assigned.
The presence or absence of partial coding regions between exons 1 and 2 of the pfhrp2 and pfhrp3 genes was assessed using two nested PCR assays as previously described [18,19] (Table 1). Positive and negative controls were used within each experiment. For the positive controls, we used DNA from previously confirmed clinical isolates available in our laboratory, which had been validated in earlier published studies and samples confirmed by sequencing in prior work. All negative samples were screened a second time and samples showing double deletions in both target regions underwent a third round of amplification.
Putative SNPs at positions 707/718/725 of the polymorphic region of domain 2 of the pfs47 gene were genotyped as previously described [15,31] (Table 1). Sample “M1” from the migrant and two local samples were sequenced.
2.4. Molecular Markers for P. vivax: pvcsp, pvmsp1-F2 block, pvmsp3α, pvmsp3β, pvs47, and pvs48/45
Four samples obtained from foreign patients with vivax malaria migrating through the country were analyzed. The subjects were recruited in the Cortés and El Paraíso departments. One of these subjects was from Afghanistan (“M2”), one from Venezuela (sample designated “M3”), and the nationality of the other two was unknown (“M4” and “M5”). Several local samples were also analyzed.
Seven protein-coding genes expressed on the surface of different P. vivax life stages were amplified and bidirectionally sequenced to assess sequence polymorphisms. For pvcsp, sequencing enabled classification between the two major types (VK210 and VK247). The pvcsp and pvmsp1 (F2) genes were amplified and sequenced as previously described [24], employing the primers detailed in Table 1. Similarly, the pvmsp3α and pvmsp3β genes were analyzed [3]. Nucleotide sequences were translated to amino acid sequences using the correct ORF, and identified haplotypes were compared against those previously reported as circulating in the country. Novel haplotypes were deposited in GenBank, with corresponding accession numbers assigned.
The full coding sequences of two genes encoding antigens proposed as potential transmission-blocking vaccine (TBV) candidates—Pvs47 and Pvs48/45—were amplified and sequenced using primers previously described [16,34,35,36].
Pvs47 was amplified by PCR in a 50 µL reaction volume containing 25 µL of 2X Taq Master Mix (Promega Corp., Madison, WI, USA), 2 µL of each primer at 10 µM (Table 1), 2 µL of BSA (10 mg/mL), and 4 µL of genomic DNA. The PCR cycling conditions consisted of an initial denaturation at 95 °C for 5 min, followed by 25 cycles of 95 °C for 1 min, 58 °C for 1 min, and 72 °C for 2 min, with a final extension at 72 °C for 10 min. A nested PCR (nPCR) was performed using 1 µL of the first-round PCR product in a 50 µL reaction containing 25 µL of Taq Master Mix, 2 µL of each nested primer at 10 µM (Table 1), and 2 µL of BSA (10 mg/mL). The program for the nested PCR included an initial denaturation at 95 °C for 5 min, followed by 35 cycles at 95 °C for 1 min, 58 °C for 1 min, and 72 °C for 2 min, with a final extension at 72 °C for 10 min. Amplicons were visualized by agarose gel electrophoresis, stained with ethidium bromide, and sequenced bidirectionally using the internal primers. A total of 23 samples from local patients and 3 from foreign migrant patients were successfully sequenced. All sequences were aligned and analyzed for the presence of polymorphisms.
The pvs48/45 gene was also amplified using several PCR reactions and under the same conditions described for pvs47 except that the annealing temperature of the second PCR was 53 °C. Primers used in the first and second reactions are listed in Table 1. Given that the gene is over 1350 nucleotides in length, its full sequence could not be obtained using only the primers from the second semi-nested PCR reaction. Therefore, nine internal primers were employed to assemble each complete sequence (Table 1, Sequencing primers). A total of 23 sequences from local isolates and 3 from migrant patients were sequenced and assembled. Sequences obtained were deposited in GenBank, with corresponding accession numbers assigned. For both pvs47 and pvs48/45, homologous sequences were deposited in GenBank and accession numbers were assigned.
3. Results
3.1. Molecular Markers for P. falciparum
Genotype and phenotype analysis was performed for two parasite genes associated with antimalarial drug resistance, pfcrt and pfmdr1. In this context, the term “phenotype” refers to the in silico translation of the gene sequences into their corresponding amino acid sequences, which allows the prediction of potential resistance-associated profiles. The pfcrt wild-type haplotype (72CVMNK76), indicating chloroquine susceptibility, was detected in all samples (n = 4), including the sample of the migrant subject named “M1”. pfmdr1 polymorphisms differed between the migrant “M1” (N86/184F/S1034/N1042/D1246) and local (N86/184F/1034C/1042D/D1246) cases, suggesting distinct genetic profiles (Table 2).
Additionally, partial coding regions of two genetic diversity markers (pfglurp and pfama1) were sequenced. For pfglurp, the migrant patient’s sample “M1” revealed a novel haplotype not previously reported in Honduras, differing by two residues from the reference sequence (GenBank accession PP681141). This new sequence was deposited in GenBank under accession PV567568. The remaining three local sequences were identical to the reference sequence PP681141. Similarly, the “M1” sample revealed a novel pfama1 haplotype, not previously reported in the country, which differed by 7 residues from the reference sequence (GenBank accession PP795732). This new haplotype was deposited in GenBank under accession number PV567569. Local samples’ sequences were identical to PP795732 (Table 2).
We also assessed the presence/absence of pfhrp2 and pfhrp3, genes encoding antigens widely used in malaria rapid diagnostic tests (RDTs). The migrant subject’s sample tested positive for both loci (pfhrp2+/pfhrp3+), whereas local samples exhibited an pfhrp2+/pfhrp3− genotype (Table 2).
Regarding pfs47, three polymorphic positions of domain 2 of the pfs47 gene (707, 718, and 725), which have been used to infer the possible geographic origin of parasite strains [31], were evaluated. “M1” exhibited the 707C/718C/725C genotype, whereas the local samples showed the T707/T718/T725 genotype (Table 2).
3.2. Molecular Markers for P. vivax
We evaluated six molecular markers (pvcsp, pvmsp1-F2 block, pvmsp3αN1 and N2, and pvmsp3βN1 and N2) commonly used to assess P. vivax genetic diversity. For pvcsp, all analyzed samples—including two from migrant individuals—exhibited four VK210-type allelic variants (“M4” and “M5”), three of which were novel haplotypes and previously unreported in Honduras (Figure 1). These new variants were deposited in GenBank (Accession No. PV567570-2) (Table 3). Notably, one sample from an Afghan subject (“M2”) revealed a VK247-type variant, which constitutes the first report of this genotype in Honduras (Accession No. PV567574).
We additionally sequenced the F2 segment of pvmsp1, encompassing one conserved block and two variable blocks (blocks 6 and 8) of the gene. Two samples collected from migrant patients (“M3” and “M5”) exhibited haplotypes previously reported in Honduras (GenBank Accession No. JQ903607 and JQ903606). In comparison, two local patient samples revealed novel haplotypes not previously described in the country (Acc. No. PV590043 and PV594050) (Table 3).
The two remaining genetic markers, pvmsp3α and pvmsp3β, were sequenced in both directions. Consensus sequences could not be generated due to the length of the sequences and the difficulty in aligning the forward primer-derived sequences with those obtained from the reverse primer. Consequently, each sequence was considered as an individual marker (pvmsp3α-N1, pvmsp3α-N2, pvmsp3β-N1, and pvmsp3β-N2). Regarding the pvmsp3α-N1 locus, two samples (“M3” and “M5”) exhibited a haplotype previously reported in the country (GenBank accession numbers PP913947-8 and PP795720). A third sample from a migrant subject (“M4”) revealed a novel haplotype, not previously documented in Honduras, which was deposited in GenBank under accession number PV590041. This haplotype showed 100% similarity with sequences reported in the Americas (e.g., AAO20877, AGR50762) and Asia (e.g., WQM98282). For the pvmsp3β-N1 locus, all three migrant samples displayed the same haplotype, which had not been previously reported in the country (Acc. No. PV590042). One local sample also revealed a novel haplotype (Acc. No. PV594049). Sequencing with the N2 primer (for both pvmsp3α and pvmsp3β) did not reveal any new haplotypes in Honduras. The three migrant patient samples all exhibited known haplotypes: pvmsp3α (Acc. No. PP913961-2) and pvmsp3β (Acc. No. PP795728-31, PP886070) (Table 3).
pvs47 and pvs48/45
The coding sequence of the pvs47 gene, comprising 1299 nucleotides, was sequenced in 23 samples from local patients and 4 from migrant subjects. Translation of this sequence yielded a 433-residue polypeptide. A single SNP was detected at position 22 of the polypeptide (F22L). Seven samples exhibited the F22 phenotype, two of which were from migrant individuals (“M2” and “M4”), while the remaining samples displayed the 22L phenotype, including “M3” and “M5”. Table 3 also describes 13 additional positions that have been reported as polymorphic in the literature [14,16,35]. GenBank accession numbers were assigned to the genotypes described in this study (Acc. No. PV700528-9, PV30102).
The pvs48/45 gene, consisting of 1350 nucleotides and encoding 450 amino acids, was also sequenced in 23 local samples and 3 from migrant individuals. Three polymorphic positions were identified: H211N, K250N, and E353Q. “M2” exhibited the H211/250N/353Q phenotype, while “M3” and “M5” revealed the 211N/K250/E353 genotype. “M4” could not be successfully sequenced. An additional nine positions previously reported as polymorphic [16] are shown in Table 3. The remaining local sequences (n = 18) revealed the haplotype E35/Y196/K211/K250/D335/E353/A376/I380/K390/K418 (Acc. No. PV700525-7).
4. Discussion
Human mobility represents one of the main challenges for infectious disease control globally. Migratory flows can facilitate the introduction of vector-borne diseases, including malaria, into regions with low endemicity or near elimination [38]. Such mobility poses the additional risk of introducing Plasmodium strains carrying drug resistance mutations or genetic variants not previously detected locally. In this context, the implementation of molecular tools for malaria surveillance is increasingly relevant, as they provide valuable information that complements conventional case surveillance and supports regional elimination goals [39].
In Honduras, malaria surveillance is coordinated by the Ministry of Health through standardized protocols in which thick blood smear microscopy remains the gold standard for case confirmation and reporting [8]. All confirmed malaria cases are entered into the national epidemiological information system, which serves as the country’s repository of case data. While microscopy is reliable and cost-effective, its limitations in detecting low-density and mixed infections highlight the potential of molecular approaches to add resolution by identifying Plasmodium species and genetic variants, and by contributing to the monitoring of drug resistance [20,40]. The present study was therefore designed not to replace, but to complement conventional malaria surveillance with molecular evidence.
In this study, six molecular markers of P. falciparum and eight of P. vivax were analyzed in samples from malaria patients in transit through Honduras en route to northern regions of the Americas, as well as in samples from local patients. The objective of characterizing these Plasmodium isolates was to evaluate the informativeness of the 14 molecular markers in distinguishing whether the infections originated from external geographic regions or within the national territory. Additionally, a detailed characterization of the genotypes circulating within the country may soon become a valuable tool for distinguishing relapses from reinfections in malaria patients treated with primaquine or tafenoquine, especially in a context of limited access to next-generation sequencing (NGS) technologies [41].
The first marker evaluated in patients with P. falciparum malaria was pfcrt, a transporter gene whose mutations at codons 72 to 76 confer resistance to chloroquine (CQ). The haplotype identified in “M1”, a sample from a migrant patient, was 72CVMNK76, described as the wild-type phenotype and associated with CQ susceptibility. CQ-resistant phenotypes (CVIET, SVMNT, CVIDT, and CVMNT) have been reported for several decades in nearly all malaria-endemic regions [42], except Central America, where the wild-type CVMNK remains the only circulating haplotype [3,20,21,43]. The fact that patient “M1” was infected with a strain carrying the wild-type pfcrt phenotype suggests that the origin of the infection may have been Honduras or a neighboring country, including Haiti or the Dominican Republic. However, the withdrawal of CQ as treatment in several African countries has led to an almost complete re-emergence of CQ-sensitive parasite populations [9,29,44,45,46,47], along with the appearance of some CQ-sensitive populations in Asia [48]. Consequently, the CVMNK haplotype can no longer be considered exclusive to the Central American region.
A second antimalarial drug resistance marker evaluated in this study was pfmdr1. The haplotype detected in “M1” was N86/184F/S1034/N1042/D1246 (NFSND haplotype), which differs at positions 1034 and 1042 from the local strains, all of which exhibited the NFCDD phenotype. Previous studies have analyzed the pfmdr1 gene in strains collected in Honduras. In 2011, the N86 phenotype was identified in 30 samples from Honduras, while one sample from Africa and one from the Pacific region showed the mutant 86Y genotype [22]. In 2020, Valdivia et al. analyzed 16 strains from Honduras that revealed the N86, 184F, D1246 haplotype [17]. In 2021, 51 local strains were analyzed, and all displayed the NFCDD haplotype [20]. On the other hand, the reference strain HB3, originally isolated in Honduras, carries the NFSDD haplotype. Thus, the NFSND haplotype had not been previously reported in Honduras and may be indicative of an infection acquired outside the national territory.
Two genetic markers widely used to estimate parasite genetic diversity were also sequenced. For both pfglurp and pfama1, “M1” exhibited haplotypes that had not been previously reported in Honduras. Recently, Zamora et al. analyzed both genes in 90 P. falciparum strains collected from malaria-endemic regions of Honduras [3]. In both cases, 89 out of the 90 strains were identical, and for pfglurp, only one strain showed a 56-nucleotide deletion, indicating high homogeneity in these markers. A GenBank search for the pfglurp genotype found in “M1” showed 100% identity with the 3D7 strain (Acc. No. MG578504) and with other entries reported from India and Indonesia (Acc. Nos. KY425859–64–95, AF191066), among others. For pfama1, the highest similarity of the genotype found in “M1” to sequences in GenBank was 98.42%. Given the low genetic diversity of the parasite population in Honduras, the detection of divergent variants in pfglurp and pfama1 in “M1” further supports the hypothesis of an imported strain from a foreign country.
Our study also screened for deletions on pfhrp2 and pfhrp3. Their presence or absence has been relevant since 2010 due to their involvement in rapid diagnostic tests (RDTs) [49]. The “M1” sample showed the presence of both gene segments (pfhrp2+/pfhrp3+), whereas six local samples displayed the pfhrp2+/pfhrp3− haplotype. Deletions at both loci have been previously investigated in Central America. The first study, published in 2015, analyzed 68 samples collected in the city of Puerto Lempira. All samples tested positive for pfhrp2, and only 50% were positive for pfhrp3 [18]. In 2018, a follow-up study was published analyzing 128 samples from Honduras, Guatemala, and Nicaragua [19]. In that study, only 8.6% of the isolates were pfhrp3+, suggesting that the pfhrp2+/pfhrp3+ haplotype is not common in Honduras, although its occurrence in the circulating parasite population cannot be entirely ruled out.
The final P. falciparum marker analyzed was pfs47, a gene that mediates the parasite’s ability to evade the mosquito immune system and is therefore critical for parasite survival and transmission [50]. It has been shown that pfs47 exhibits strong geographic population structure, with distinct haplotypes predominating across continents [31,41]. The major differences among these haplotypes are based on polymorphisms at nucleotide positions 707, 718, 725, and 742. Sequencing of the gene in sample “M1” revealed the genotype 707C/725C (CC), whereas local samples in this study and 31 additional samples analyzed in a previous study [15] displayed the 707T/725T (TT) genotype. The TT genotype has been described as the most frequent in Latin America, while the CC genotype is more common in Africa. This evidence supports the hypothesis of an infection acquired outside the country.
We also analyzed four molecular markers commonly used to assess P. vivax genetic diversity—pvcsp, pvmsp1 (F2 block), pvmsp3α, and pvmsp3β—in both local and migrant patient samples. All pvcsp sequences belonged to the VK210 type, except one from an Afghan migrant (“M2”), which revealed the VK247 genotype, marking its first report in Honduras, supporting the hypothesis that this isolate may have a non-American origin. Among the VK210 variants, three novel haplotypes were identified compared to those previously described in a study that sequenced 84 samples collected from all malaria-endemic regions at that time [24]. Sequencing of the pvmsp1-F2 segment revealed two migrant samples (“M3” and “M5”) with known Honduran haplotypes, while two local samples exhibited previously undescribed variants [24], suggesting locally acquired infections. Unfortunately, pvmsp1 could not be sequenced in “M2” or “M4”.
At the pvmsp3α-N1 locus, two migrant samples carried known haplotypes [3], while a third (“M4”) displayed a novel variant with 100% similarity to sequences from South America (Acc. No. AAO20877, AGR50762) and Myanmar (Acc. No. WQM98282). This result, along with the novel VK210 haplotype found for pvcsp, would also suggest that this parasite strain could have been imported. For pvmsp3β-N1, all three migrant samples and one local sample revealed previously unreported haplotypes [3]. Thus, this locus does not allow for discrimination of the parasites’ origin.
Finally, the complete coding sequences of two candidate transmission-blocking vaccine genes, pvs47 and pvs48/45, were sequenced. In pvs47, only a single SNP was detected, resulting in a phenylalanine at residue 22 in samples “M2” and “M4,” whereas local samples and the other two migrant samples displayed the 22L phenotype. For pvs48/45, three polymorphic positions were identified: H211N, K250N, and E353Q. Sample “M2” exhibited the H211/250N/353Q phenotype, while “M3” and “M5” showed the 211N/K250/E353 genotype. “M4” could not be successfully sequenced. Eighteen local samples revealed a diverse haplotype (K211/K250/E353). These findings strongly suggest that both markers are highly informative for inferring the geographic origin of P. vivax strains, particularly in the current context of low transmission and reduced genetic diversity in Honduras.
Taken together, these results demonstrate the value of incorporating molecular markers as a complementary tool to the national malaria surveillance system. By situating the findings in the broader problem of imported diseases, we emphasize that molecular approaches add resolution to case detection, help identify imported strains with potential drug resistance, and provide data on parasite population structure. These contributions are particularly relevant in countries like Honduras, where malaria transmission is now focalized, and where elimination goals will depend on distinguishing local from imported cases with greater accuracy.
5. Conclusions
This study fulfilled its objective of assessing the utility of multiple molecular markers to differentiate between locally acquired and potentially imported malaria infections in Honduras, a country currently experiencing low transmission and limited genetic diversity of the parasites. By analyzing six P. falciparum and eight P. vivax genetic markers, we identified novel haplotypes and allelic variants that had not been previously reported in the country, particularly in samples from migrant patients. Some markers—such as pfmdr1, pfama1, pfs47 for P. falciparum, and pvcsp, pvmsp3α, and the transmission-blocking vaccine candidates pvs47 and pvs48/45 for P. vivax—proved especially informative in inferring the likely geographic origin of infections. These findings demonstrate the value of molecular surveillance in detecting the introduction of foreign strains and complementing conventional surveillance systems and in guiding public health strategies, especially in settings where advanced sequencing technologies remain unavailable. We also acknowledge the main limitations of the study, including the small number of migrant samples and the lack of systematic follow-up of clinical outcomes, which should be addressed in future research with larger cohorts.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization World Malaria Report 2024 Global Malaria Programme (GMP), WHO Geneva, Switzerland 2024316
- 2Pinto A. Archaga O. Mejia A. Escober L. Henriquez J. Montoya A. Valdivia H.O. Fontecha G. Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran-Nicaraguan Border Pathogens 202110143210.3390/pathogens 1011143234832588 PMC 8617645 · doi ↗ · pubmed ↗
- 3Zamora A. Pinto A. Escobar D. Valdivia H.O. Chaver L. Ardon G. Carranza E. Fontecha G. Genetic diversity of Plasmodium vivax and Plasmodium falciparum field isolates from Honduras in the malaria elimination phase Curr. Res. Parasitol. Vector Borne Dis.2025710023010.1016/j.crpvbd.2024.10023039759387 PMC 11699087 · doi ↗ · pubmed ↗
- 4Agudelo Higuita N.I. Franco-Paredes C. Henao-Martinez A.F. Mendez Rojas B. Suarez J.A. Naranjo L. Alger J. Migrants in transit across Central America and the potential spread of chloroquine resistant malaria-a call for action Lancet Reg. Health Am.20232210050510.1016/j.lana.2023.10050537214770 PMC 10193226 · doi ↗ · pubmed ↗
- 5Loyola-Cruz M.A. Duran-Manuel E.M. Cruz-Cruz C. Bravata-Alcantara J.C. Gutierrez-Munoz V.H. Marquez-Valdelamar L.M. Leal-Escobar B. Vasquez-Jimenez E. Cureno-Diaz M.A. Lugo-Zamudio G.E. Imported malaria cases by Plasmodium falciparum and Plasmodium vivax in Mexican territory: Potential impact of the migration crisis Travel Med. Infect. Dis.20246210277310.1016/j.tmaid.2024.10277339393476 · doi ↗ · pubmed ↗
- 6Fontecha G. The Honduran diaspora and infectious diseases: An urgent need for action Travel Med. Infect. Dis.20235310256710.1016/j.tmaid.2023.10256736958705 · doi ↗ · pubmed ↗
- 7He X. Zhong D. Zou C. Pi L. Zhao L. Qin Y. Pan M. Wang S. Zeng W. Xiang Z. Unraveling the Complexity of Imported Malaria Infections by Amplicon Deep Sequencing Front. Cell. Infect. Microbiol.20211172585910.3389/fcimb.2021.72585934595134 PMC 8477663 · doi ↗ · pubmed ↗
- 8Secretaría de Salud de Honduras Norma Nacional de Malaria de Honduras Secretaría de Salud de Honduras Tegucigalpa, Honduras 2018