Beyond TFRC: The Pivotal Role of mGluR2 in Feline Calicivirus Entry and Replication
Ruibin Qi, Hongtao Kang, Yupeng Yang, Kexin Feng, Zhe Liu, Silu Gao, Qian Jiang, Liandong Qu, Jiasen Liu

TL;DR
This study reveals that mGluR2 is a new host protein involved in feline calicivirus entry, independent of TFRC, offering insights into viral infection mechanisms.
Contribution
The study identifies mGluR2 as a novel, independent host factor for FCV entry, distinct from TFRC.
Findings
mGluR2 interacts directly with FCV and enhances viral entry into host cells.
Reduced mGluR2 levels significantly decrease FCV infectivity, even when TFRC is present.
mGluR2 influences endocytic vesicle formation and F-actin polymerization during FCV infection.
Abstract
Feline calicivirus (FCV) is a prevalent virus that induces respiratory and oral diseases in felines and serves as a valuable model for investigating other challenging caliciviruses, such as human norovirus. In previous research, we identified a cellular protein known as transferrin receptor (TFRC) that facilitates the entry of FCV into feline cells; however, we sought to determine whether additional proteins are implicated in this process. To explore this, we examined the role of another protein, metabotropic glutamate receptor 2 (mGluR2), in the context of FCV infection through laboratory techniques designed to assess protein interactions and cellular infection. Our results demonstrated that mGluR2 directly interacts with FCV and enhances the virus’s entry into host cells. Notably, when mGluR2 levels were diminished, FCV exhibited significantly reduced infectivity, even in the presence…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
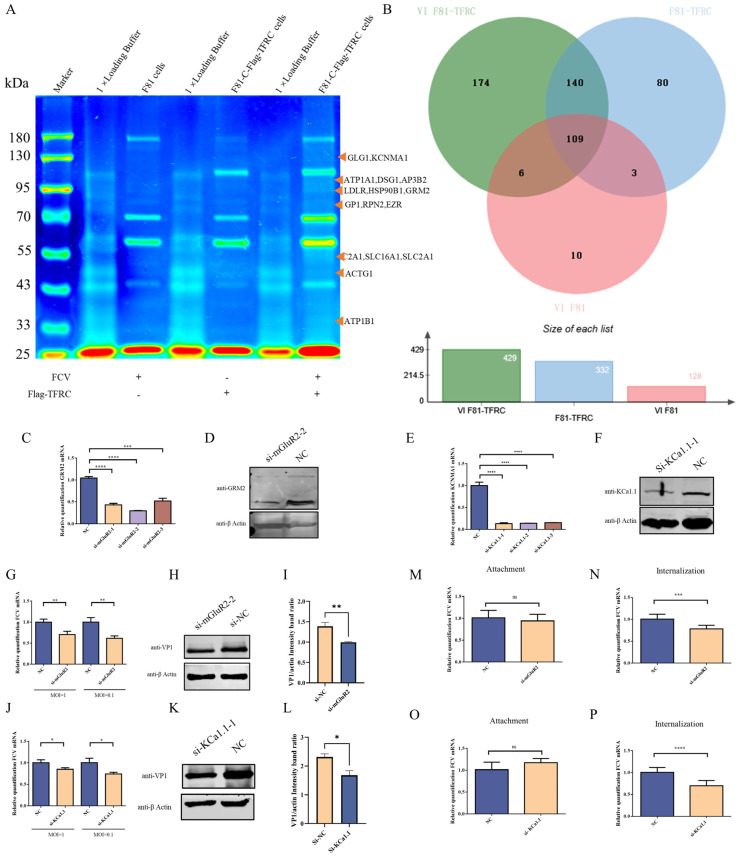 Figure 1
Figure 1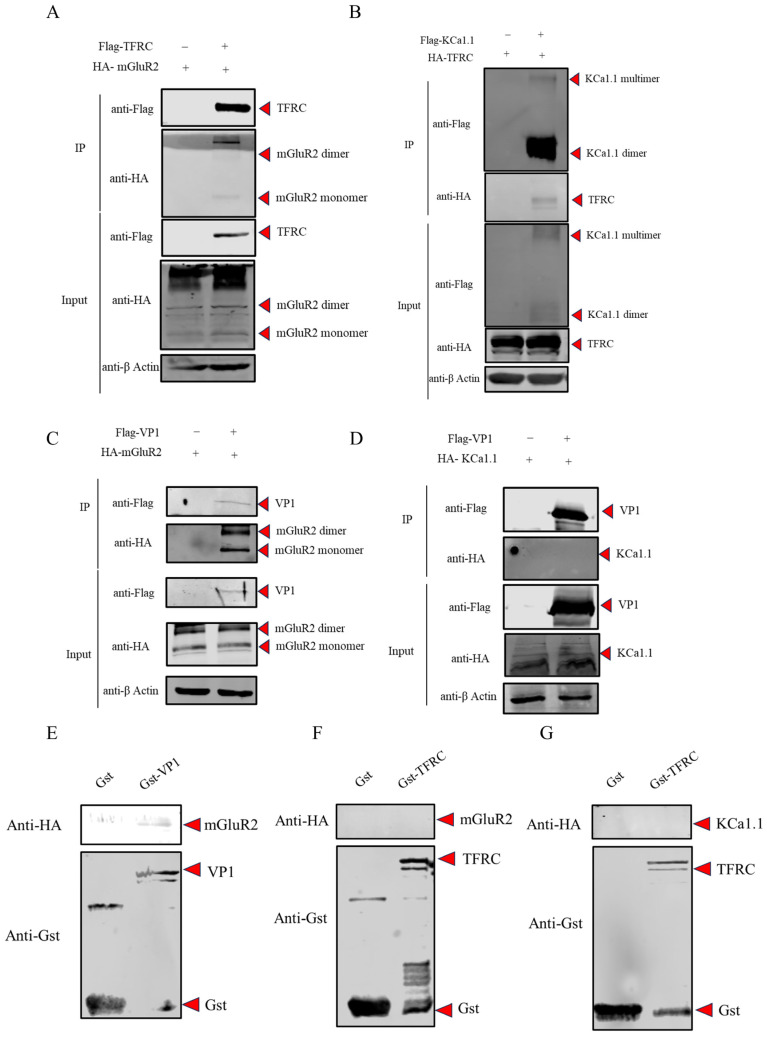 Figure 2
Figure 2 Figure 3
Figure 3- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsViral gastroenteritis research and epidemiology · Animal Virus Infections Studies · Viral Infections and Immunology Research
1. Introduction
Receptors are the key for viruses to enter into the host cells and the studies of the interaction between viruses and receptors not only help to understand the pathogenic mechanism of viruses but also facilitate the prevention of viral infections in hosts [1,2]. The Caliciviridae family comprises important pathogens with a broad host range and most members of Caliciviridae have no suitable platform for in vitro culture which greatly restrict the studies of viral receptors participating in calicivirus infection [3,4,5]. Numerous studies have demonstrated the diversity of viral receptors among different genera within the Caliciviridae family. For instance, Caliciviruses can interact with various carbohydrates, including histo-blood group antigens (HBGAs), sialic acid, and heparan sulfate proteoglycans, as well as proteins such as CD300lf and JAM-A, to facilitate cellular binding [6,7,8,9]. The clathrin-mediated endocytosis (CME) pathway is the primary mechanism by which Caliciviruses enter host cells following attachment [10]. However, the precise mechanisms by which Caliciviruses regulate the CME pathway remain to be elucidated.
As one of the very few caliciviruses that can efficiently replicate in vitro, Feline calicivirus (FCV) offers unique advantages for basic research on the Caliciviridae family. The exploration of FCV receptors could help control the FCV infection and successful culture of other caliciviruses in vitro. Junctional adhesion molecule A (JAM-A) serves as the functional receptor for FCV and facilitates the attachment of the virion to host cells [11,12]. And the knockout of JAM-A only has impact on the attachment of FCV to cells which indicates other host factors expert JAM-A mediating the internalization of FCV. Our previous research demonstrated that TFRC can directly interact with FCV VP1 and plays a role in the internalization stage of FCV. However, knocking out TFRC only inhibited, but did not completely block, the entry of FCV into F81 cells. This led us to hypothesize that FCV utilizes multiple host factors as internalization receptors. Furthermore, it remains to be determined whether other host factors are involved in TFRC-mediated FCV internalization.
Metabotropic glutamate receptor 2 (mGluR2), a member of the G protein-coupled receptor (GPCR) family, plays a pivotal role in the regulation of glutamatergic neural signaling. Previous studies have elucidated its involvement in viral entry by modulating membrane fluidity during the endocytosis of influenza viruses, coronaviruses, and other viral entities, facilitated by TFRC [13,14]. As a crucial downstream signaling molecule of mGluR2, the calcium-activated potassium channel subunit alpha-1 (KCa1.1) is capable of modulating intracellular calcium ion concentrations to regulate the activity of endocytosis-related proteins, such as F-actin, thereby mediating the internalization stage of viral entry [14].
In this study, we demonstrated that mGluR2 can also mediate the internalization of FCV in F81 cells by influencing the activity of KCa1.1, in addition to TFRC. Moreover, mGluR2 can cooperate with KCa1.1 to facilitate FCV entry into host cells independently of TFRC. Additionally, mGluR2 was found to specifically interact with both VP1 and TFRC. Collectively, these findings provide further insights into the interactions between FCV virions and viral receptors, including mGluR2 and TFRC. These findings not only enhance our understanding of the molecular mechanism underlying “attachment-endocytosis” in FCV by elucidating the ability of caliciviruses to adapt to host cells through “multi-receptor coordination,” thereby offering a framework for comprehending receptor selection in other challenging-to-culture caliciviruses such as noroviruses; but also identify novel targets for the prevention and control of FCV.
2. Material and Methods
2.1. Cell Culture and Virus
The F81, F81-c-Flag-TFRC, F81-TFRC^−/−^ and HEK293T cell lines were maintained at 37 °C in a 5% CO2 atmosphere using Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Grand Island, NY, USA, Catalog No. C11995500CP) supplemented with 10% fetal bovine serum and 1% penicillin (100 U/mL) and streptomycin (100 μg/mL). The F81 cell line (RRID: CVCL_9259) was obtained in 2023 from the Kunming Cell Bank of the Chinese Academy of Sciences (KCB) and has been confirmed to be free from mycoplasma contamination, as well as authenticated through species identification. The FCV 2280 strain was preserved at the Harbin Veterinary Research Institute.
2.2. Reagents and Antibodies
The study utilized various antibodies and reagents, including the monoclonal anti-Flag^®^ M2 antibody produced in mouse (Sigma-Aldrich, St. Louis, MO, USA, Catalog No. F1804), HA tag polyclonal antibody produced in rabbit (Proteintech Group, Inc., Rosemont, IL, USA, Catalog No. 51064-2-AP), rabbit monoclonal antibody against mGluR2 (Thermo Fisher, Waltham, MA, USA, Catalog No. 702952), and KCa1.1 polyclonal antibody produced in rabbit (Abmart, Shanghai, China, Catalog No. TD8570). Additionally, the study employed anti-mouse IgG (H+L) antibody, Human Serum Adsorbed, DyLight^TM^ 800-Labeled (SeraCare, Milford, MA, USA, Catalog No. 5230-0415), and anti-rabbit IgG (H+L) antibody, DyLight^TM^ 800-Labeled (SeraCare, Milford, MA, USA, Catalog No. 5230-0412).
2.3. Plasmid Constructs
The pCMV-HA-C and pcaggs-C-Flag vectors were utilized to construct mammalian expression systems, while the pGEX-4T-1 vector was employed to facilitate protein expression in competent E. coli BL21-CodonPlus (DE3) cells. The gene sequences for feline mGluR2 (XM_006928832.5) and KCa1.1 (XM_011287302.4) were amplified using reverse transcription polymerase chain reaction (RT-PCR) from an F81 cell DNA library.
2.4. The Potential TFRC-Binding Proteins Identified by Co-IP Followed by LC-MS Assays
After a 24 h culture period in 10 cm^2^ dishes, F81 and F81-c-Flag-TFRC cells were infected with the FCV 2280 strain at a multiplicity of infection (MOI) of 10. Three hours post-infection, host proteins from both FCV-infected and F81-c-Flag-TFRC cells were extracted using 500 μL of NP-40 lysis buffer to conduct a Co-immunoprecipitation (Co-IP) assay with 20 μL of Anti-Flag^®^ M2 Magnetic Beads (Sigma-Aldrich, St. Louis, MO, USA, Catalog No. M8823). The Co-IP complexes were washed with 500 μL of NP40 with three times and analyzed with Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) to identify the distinct protein bands present in F81 cells infected with FCV 2280 compared to F81-c-Flag-TFRC cells infected with FCV or F81-c-Flag-TFRC cells infected with PBS. Subsequently, the unique protein bands were characterized through Liquid Chromatograph Mass Spectrometer (LC-MS) analyses.
For the detection of proteins in gels utilizing LC-MS, proteins are initially digested with trypsin. The resulting peptides are then separated using an Easy nLC system (Thermo Fisher Scientific, Waltham, MA, USA, Catalog No. 1200), which is equipped with a Thermo Acclaim PepMap RSLC column. The separation employs mobile phases consisting of 0.1% formic acid in water and 0.1% formic acid in 80% acetonitrile, with elution occurring at a flow rate of 300 nL/min according to a specified gradient. Detection is performed using a Q Exactive HF-X (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer. Subsequent protein identification is achieved through data processing with Proteome Discoverer 2.2 software, in conjunction with a MASCOT 2.6 database search, ensuring a false discovery rate (FDR) of less than 0.01.
2.5. Small-Interfering RNA (siRNA) Transfection
A 30 pmol quantity of Small interfering RNAs (siRNAs) targeting mGluR2 and KCa1.1, along with scrambled siRNA controls, was obtained from GenePharma Co., Ltd. (Suzhou, China) and subsequently transfected into cells using 6 μL of the siRNA-mate plus reagent, in accordance with the manufacturer’s protocol.
2.6. Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA was isolated from the cells utilizing the AxyPrep Multisource Total RNA Kit (AxyPrep, Union City, CA, USA, Catalog No. AP-MN-MS-RNA) and was reverse transcribed into complementary DNA (cDNA) using the PrimeScript^TM^ RT Reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, Otsu, Shiga, Japan, Catalog No. RR047A). Quantitative real-time PCR was performed on a QuantStudio 5 Fluorescent Quantitative PCR Instrument (Applied Biosystems, Waltham, MA, USA), employing the ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech Co., Nanjing, Jiangsu, China, Catalog No. Q711). The primer sequences used in the analysis are provided in Table S1.
2.7. Western Blotting (WB)
After treatment, the cells were lysed in each well using 200 μL of RIPA lysis buffer (Beyotime Biotechnology, Haimen, Jiangsu, China, Catalog No. P0013B). The total cell lysates were then subjected to boiling following the addition of loading buffer to prepare for SDS-PAGE electrophoresis. Primary antibodies, diluted according to the specified protocols, were incubated for 16 h. Subsequently, secondary antibodies (Goat anti-Rabbit or Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody), diluted at a ratio of 1:10,000, were incubated for 1 h. Image visualization was performed using an Infrared Imaging System (LI-COR, Lincoln, NE, USA, Catalog No. Odyssey CLX).
2.8. Viral Infection
After a 48 h culture period, the cells were infected with the FCV 2280 strain at various multiplicities of infection (MOIs). The entire cell population was harvested 12 h post-infection to evaluate VP1 expression through qRT-PCR and WB assays.
2.9. Measurement of Attachment and Internalization Rates
For virus attachment studies, the cells were infected with FCV 2280 at an MOI of 10 and incubated at 4 °C for 1 h. Following synchronized adsorption for 1 h, unbound FCV virions were removed by washing with Phosphate-Buffered Saline (PBS), and the relative levels of bound FCV virions were quantified using qRT-PCR. To assess internalization rates, cells with bound FCV were incubated at 37 °C, washed with acidic PBS (pH 2.5) to eliminate any remaining attached FCV virions, and subsequently analyzed by qRT-PCR.
2.10. Co-Immunoprecipitation (Co-IP) Analysis
Following a 24 h incubation period in a 6-well plate, F81 cells were co-transfected with 2 μg of HA/Flag-tagged eukaryotic expression vectors or blank control vectors. Forty-eight hours post-transfection, the F81 cells were lysed using 200 μL of NP-40 to extract total proteins for subsequent co-immunoprecipitation (Co-IP) analysis with 20 μL of Anti-Flag^®^ M2 Magnetic Beads (Sigma-Aldrich, St. Louis, MO, USA, Catalog No. M8823). The Co-IP complexes underwent three washes with 500 μL of NP-40 prior to Western blot (WB) analysis.
2.11. Glutathion-S-Transferase (GST) Pull-Down Assay
Recombinant proteins Gst-2280 VP1, Gst-TFRC, and Gst were expressed and purified using BeyoMag^TM^ GSH Magnetic Agarose Beads (Beyotime, Haimen, Jiangsu, China, Catalog No. P2258). After purification and dialysis, 25 μg of each recombinant protein was incubated with 25 μL of BeyoMag™ GSH Magnetic Agarose Beads to perform Gst pull-down assays. Subsequently, total cellular proteins or Flag-tagged mGluR2 and KCa1.1 proteins were extracted from F81 cells and incubated with the GSH beads conjugated to the recombinant proteins. Following an overnight incubation at 4 °C, the pull-down complexes were washed with NP-40 and analyzed via WB analysis.
2.12. F-Actin Polymerization Assay
F-actin, or filamentous actin, constitutes a fundamental element of the cytoskeleton. It is generated through the dynamic polymerization of G-actin, which are globular actin monomers, and exhibits the reversible characteristic of “assembly–disassembly”. Through its dynamic structural modifications, F-actin facilitates the spatiotemporal precision and efficiency of the endocytosis process [15,16]. To detect the F-actin polymerization rates, the cells were treated and infected with FCV 2280 at an MOI of 10 and incubated at 37 °C for 30 min to assess F-actin polymerization rates. At 30 min post-infection, the cells were fixed with 4% paraformaldehyde and subsequently washed three times with PBS. The cells were then incubated with phalloidin iFluor^TM^ 488 for 1 h. The viral internalization assay was examined utilizing a Confocal Laser Scanning Microscope (ZEISS, Oberkochen, Baden-Württemberg, Germany, Catalog No. LSM980) equipped with Fast Airyscan technology to assess F-actin polymerization rates. Post-acquisition of confocal images, the fluorescence intensity of F-actin was quantified using Image J software (version 1.8.0, National Institutes of Health, Bethesda, MD, USA) in accordance with the standard intensity analysis protocol. Regions of interest (ROIs) were meticulously selected to minimize background interference, and the mean fluorescence intensity within each ROI was calculated for subsequent statistical analysis.
2.13. Statistics
Statistical analyses were conducted using the student’s t-test and one-way analysis of variance (ANOVA) with GraphPad Prism software (version 10). Statistical significance was defined as p-values less than 0.05, indicating significance, and p-values less than 0.01, indicating high significance.
3. Results
3.1. Identification of Host Proteins Interacting with TFRC
Our prior investigations have demonstrated that TFRC is integral to the internalization phase of the FCV life cycle through its direct interaction with the FCV structural protein VP1. Nonetheless, the involvement of additional proteins in this process remains to be elucidated. To investigate this, a Co-IP assay was conducted to identify potential host proteins interacting with TFRC in F81 cells, under both FCV-infected and uninfected conditions. The results indicated that 140 host proteins commonly interact with TFRC, while 174 host proteins uniquely associate with TFRC during FCV infection (Figure 1A,B) (Original dates were in Supplementary Materials Table S2). Notably, among these proteins, mGluR2, HSPA5, LDLR, and ITGB2 have been implicated in facilitating the entry stage of various viruses, such as SARS-CoV-2, Porcine Epidemic Diarrhea Virus (PEDV), and Crimean-Congo Hemorrhagic Fever Virus (CCHFV).
3.2. mGluR2 and KCa1.1 Modulate the Replication of FCV by Regulating Its Internalization into Host Cells
To assess the influence of mGluR2 on FCV replication, three distinct siRNAs specifically targeting mGluR2 were designed and synthesized to suppress mGluR2 protein expression in F81 cells. The results demonstrated that the siRNAs effectively down-regulated both mRNA and protein expression levels of mGluR2 (Figure 1C,D). Consequently, the levels of VP1 protein and viral cDNA copies were significantly diminished following the reduction in intracellular mGluR2 protein expression (refer to Figure 1G–I). To further elucidate the role of mGluR2 in FCV entry, additional analyses were performed. The findings indicated that mGluR2 facilitates the internalization of the virus into cells without influencing viral attachment (Figure 1M,N).
During influenza virus infection, mGluR2 could modulate the activity of KCa1.1, thereby influencing the internalization stage of the virus by affecting F-actin polymerization rates. To explore whether a comparable mechanism is involved in the entry of FCV into host cells, the mRNA and protein expression levels of KCa1.1 in F81 cells were effectively downregulated using siRNA targeting KCa1.1 (Figure 1E,F). This downregulation of KCa1.1 expression subsequently inhibited FCV replication, as evidenced by qRT-PCR and WB analyses (Figure 1J–L). Similar to the role of mGluR2, KCa1.1 appears to influence the internalization rather than the attachment phase of FCV infection (Figure 1O,P). Collectively, these findings indicate that mGluR2 may collaborate with TFRC to facilitate the internalization of FCV by modulating KCa1.1 activity.
3.3. The Interaction Analysis Among mGluR2, KCa1.1, TFRC and FCV VP1
To gain a more comprehensive understanding of the molecular mechanisms underlying the interaction between mGluR2 and the FCV entry stage, we employed Co-IP assays to investigate the interactions among mGluR2, FCV VP1, TFRC, and KCa1.1. The results indicated that TFRC interacts with both mGluR2 and KCa1.1 (Figure 2A,B), consistent with the findings from LC-MS assays identifying potential host protein interactions with TFRC. However, only mGluR2 was found to interact with FCV VP1 (Figure 2C,D), suggesting that mGluR2 may function as an entry receptor for FCV.
To further investigate whether mGluR2 could serve as an internalization receptor during FCV infection, GST pull-down assays were conducted to assess the direct interactions between mGluR2 and FCV VP1, as well as between mGluR2 or KCa1.1 and TFRC. The assays revealed that only mGluR2 directly interacts with FCV VP1 (Figure 2E), while TFRC does not directly interact with either mGluR2 or KCa1.1 (Figure 2F,G). These findings suggest that mGluR2 collaborates with KCa1.1 to mediate FCV internalization, a process that does not necessitate the involvement of TFRC.
3.4. mGluR2 Mediates FCV Internalization Without TFRC Involved
Previous research has demonstrated that TFRC is essential for agonist-induced endocytosis of mGluR2. To investigate whether a similar mechanism is present during FCV infection, siRNA targeting mGluR2 and KCa1.1 was transfected into F81-TFRC^−/−^ cells to assess the role of TFRC in FCV internalization mediated by mGluR2 and KCa1.1. The results indicated that downregulation of mGluR2 or KCa1.1 protein expression inhibited FCV replication or internalization in F81 cells lacking TFRC expression (Figure 3A,C,D,F), consistent with observations in F81 cells. Furthermore, the attachment rates of FCV virions in F81-TFRC^−/−^ cells were not affected by modulation of mGluR2 or KCa1.1 protein expression (Figure 3B,E). Consequently, we propose that mGluR2 and KCa1.1 independently modulate the FCV entry process without reliance on TFRC.
To further validate our conclusions, phalloidin iFluor^TM^ 488 was employed to assess the regulation of F-actin polymerization rates by FCV infection and siRNA interference. The results demonstrated that FCV infection led to an increase in F-actin polymerization rates in F81 cells, regardless of whether they were transfected with control siRNA (Figure 3G,H,K,L). However, knockdown of TFRC did not significantly affect the FCV-induced increase in F-actin polymerization rates (Figure 3I). Furthermore, the reduction in mGluR2 protein expression inhibited the F-actin polymerization induced by FCV infection (Figure 3M). These findings suggest that mGluR2 facilitates FCV internalization by enhancing the activity of KCa1.1, thereby promoting F-actin polymerization.
4. Discussion
The clathrin-mediated endocytosis (CME) pathway is a ubiquitous mechanism employed by a wide variety of viruses, including feline calicivirus (FCV), to gain entry into host cells [10,16,17,18]. The formation of clathrin-coated vesicles (CCVs) represents a crucial initial phase of CME, which can occur spontaneously or be induced by viral infections, such as those caused by the influenza virus and reovirus [19,20,21]. The majority of CME processes necessitate the involvement of the Adaptor Protein 2 (AP2) complex, with the YXXΦ motif serving as a pivotal functional domain that facilitates interactions between the AP2 complex and other associated proteins [22,23]. Viruses such as Hepatitis B Virus (HBV) and White Spot Syndrome Virus (WSSV), whose structural proteins contain the YXXΦ motif, can directly engage with the AP2 complex to initiate CME [24,25]. Conversely, viruses lacking the YXXΦ motif, such as FCV, can interact with host membrane receptor proteins to recruit the AP2 complex and trigger CME [26].
In this study, we identified another FCV entry receptor, mGluR2, besides TFRC and verified mGluR2 could specifically interact with FCV VP1 and TFRC. To further exploit the mechanisms of mGluR2 mediating FCV internalization, KCa1.1 was also down-regulated by siRNA, and we found it influences FCV replication by mediating viral internalization stage and could not interact with FCV VP1. This indicated KCa1.1 is likely to be regulated by mGluR2 to effect FCV entry instead of directly interacting with FCV VP1. The mechanism by which KCa1.1 regulate viral entry has been demonstrated in Influenza virus infection [14]. And our studies also showed that FCV can use a similar mechanism, as evidenced by the finding that down-regulation of mGluR2 can inhibit the F-actin polymerization rates induced by FCV infection.
It has already been confirmed that TFRC can cooperate with mGluR2 to mediate the internalization of various enveloped viruses such as Rabies Virus and SARS-CoV-2 [13,27]. Most GPCR family members undergo endocytosis through the CME pathway [28,29]. Additionally, mGluR2 could directly interact with TFRC by Gst pull-down analysis and co-localize with TFRC in early and late endosomes [13]. In our study, we did not observe a direct interaction between feline mGluR2 and TFRC, as evidenced by the detection of only weak band signals in the Co-IP analysis. The presence of a faint mGluR2-TFRC band in the Co-IP assay suggests that the interaction may be indirect, potentially mediated by intermediary proteins. Typically, direct interactions result in the formation of stable complexes that produce strong signals, whereas indirect interactions are characterized by multi-component complexes that are susceptible to dissociation during lysis and washing procedures due to their weaker affinity. This dissociation likely leads to reduced precipitate formation and subsequently weaker signals. The observed phenomenon may indicate a level of regulatory complexity, wherein intermediary proteins modulate the dynamics of mGluR2-TFRC interactions to prevent excessive binding and optimize the efficiency of viral internalization and replication. Furthermore, the knockout of TFRC did not affect the role of mGluR2 in FCV entry, suggesting that mGluR2 modulates FCV entry independently of TFRC, a mechanism that appears to differ from those observed in other viruses.
This study identifies mGluR2 and KCa1.1 as novel therapeutic targets for FCV inhibition, given their specificity for the internalization step—a critical vulnerability in the viral life cycle. Further work is needed to: (1) map the interacting domains of mGluR2 and VP1 to enable the design of blocking peptides; (2) define the signaling cascade linking mGluR2 activation to KCa1.1-mediated F-actin polymerization; and (3) explore whether other caliciviruses (e.g., noroviruses) utilize similar mGluR2-KCa1.1-dependent entry mechanisms, which would highlight a conserved viral strategy.
5. Conclusions
In summary, our research elucidates a novel entry pathway for feline calicivirus (FCV) that operates independently of transferrin receptor protein 1 (TFRC) and is mediated by the metabotropic glutamate receptor 2 (mGluR2) and large conductance calcium-activated potassium channel (KCa1.1) axis. This discovery significantly enhances our comprehension of host–virus interactions in caliciviruses and highlights mGluR2 and KCa1.1 as potential therapeutic targets for the prevention and control of FCV. Future investigations will aim to map the interaction domain between mGluR2 and the viral protein VP1, delineate the complete signaling cascade that connects mGluR2 to F-actin polymerization, and examine the conservation of this pathway in other caliciviruses, such as noroviruses.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Robilotti E. Deresinski S. Pinsky B.A. Norovirus Clin. Microbiol. Rev.20152813416410.1128/CMR.00075-1425567225 PMC 4284304 · doi ↗ · pubmed ↗
- 2Skehel J.J. Wiley D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin Annu. Rev. Biochem.20006953156910.1146/annurev.biochem.69.1.53110966468 · doi ↗ · pubmed ↗
- 3Abouelkhair M.A. Kennedy M. Caliciviridae Veterinary Microbiology John Wiley & Sons, Inc.Hoboken, NJ, USA 2022543551
- 4Cheng C. Cai X. Li J. Zhang X. Xie Y. Zhang J. In Vitro Culture of Human Norovirus in the Last 20 Years Biomedicines 202412244210.3390/biomedicines 1211244239595008 PMC 11592199 · doi ↗ · pubmed ↗
- 5Desselberger U. Caliciviridae Other Than Noroviruses Viruses 20191128610.3390/v 1103028630901945 PMC 6466229 · doi ↗ · pubmed ↗
- 6Haga K. Fujimoto A. Takai-Todaka R. Miki M. Doan Y.H. Murakami K. Yokoyama M. Murata K. Nakanishi A. Katayama K. Functional receptor molecules CD 300lf and CD 300ld within the CD 300 family enable murine noroviruses to infect cells Proc. Natl. Acad. Sci. USA 2016113 E 6248 E 625510.1073/pnas.160557511327681626 PMC 5068309 · doi ↗ · pubmed ↗
- 7Marionneau S. Ruvoën N. Le Moullac-Vaidye B. Clement M. Cailleau-Thomas A. Ruiz-Palacois G. Huang P. Jiang X. Le Pendu J. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals Gastroenterology 20021221967197710.1053/gast.2002.3366112055602 PMC 7172544 · doi ↗ · pubmed ↗
- 8Tamura M. Natori K. Kobayashi M. Miyamura T. Takeda N. Genogroup II noroviruses efficiently bind to heparan sulfate proteoglycan associated with the cellular membrane J. Virol.2004783817382610.1128/jvi.78.8.3817-3826.200415047797 PMC 374263 · doi ↗ · pubmed ↗