Hemophagocytic Lymphohistiocytosis due to Brucellosis in a Xeroderma Pigmentosum Pediatric Patient: A Case Report and Review of the Literature
Reem Shihab, Sultan Mosleh, Muhammad Takhman, Fadi Yousef, Marian Salim, Asala Abuabed, Sara Abueisheh, Mohammad Abed

TL;DR
A child with xeroderma pigmentosum developed a rare immune disorder due to a Brucella infection, marking the first known case of this combination.
Contribution
This is the first reported case of Brucella-induced hemophagocytic lymphohistiocytosis in a patient with xeroderma pigmentosum.
Findings
A pediatric patient with xeroderma pigmentosum developed hemophagocytic lymphohistiocytosis due to Brucella infection.
This case highlights the rare intersection of a genetic disorder and a specific infectious trigger for HLH.
The report emphasizes the need for awareness of atypical infections in immunocompromised patients with genetic conditions.
Abstract
We report a pediatric patient with xeroderma pigmentosum (XP) who developed hemophagocytic lymphohistiocytosis (HLH) secondary to Brucella infection—an exceedingly rare occurrence. XP is a rare autosomal recessive genetic disorder characterized by extreme ultraviolet radiation (UVR) sensitivity due to the inability to repair DNA pyrimidine dimers caused by UV exposure. This defect leads to a markedly increased risk of skin cancer and progressive neurological degeneration (Leung, 2022). HLH is a rare, potentially fatal hypersensitivity syndrome characterized by excessive activation and impaired downregulation of T-lymphocytes and macrophages. This dysregulation results in an overproduction of proinflammatory cytokines, destruction of blood cells, and subsequent tissue and organ damage (Fisman, 2000). While secondary HLH may follow various infections, Brucella-induced HLH is rare (Wolska,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
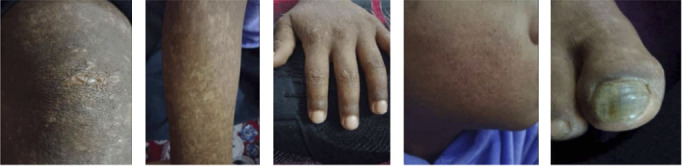 Figure 1
Figure 1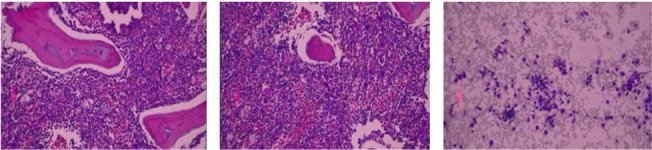 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Inflammasome and immune disorders · Dermatological and COVID-19 studies
1. Introduction
In this report, we present an unusual case of an 8 year-old female patient with XP, who exhibited prolonged and recurrent fever, abdominal and back pain, arthralgia, splenomegaly (15 cm), and pancytopenia. She was diagnosed with secondary HLH triggered by brucellosis, an association rarely reported in the literature. Early recognition of Brucella as a risk of HLH, especially in patients with underlying genetic diseases, can guide physicians to detect the condition as soon as possible and initiate life-saving interventions.
Xeroderma pigmentosum (XP) is an autosomal recessive genetic disorder characterized by deficient DNA repair mechanisms, leading to DNA damage upon ultraviolet radiation (UVR) exposure [1]. It has a variable incidence around the world, ranging from (1–45) per million live births, with a higher incidence in countries where consanguineous marriages are common [1]. Eight genetic mutations have been identified as a causative factor for XP. The first seven (XPA, XPB, XPC, XPD, XPE, XPG, and XPV) are responsible for nucleotide excision repair (NER) that is required in UVR-induced photoproduct repair, while XPV mutation causes a defect in replicating DNA with UVR damage [2].
It is characterized by photosensitivity, freckle-like pigmentations on all exposed skin, which usually starts before 2 years of age, and high susceptibility to skin cancers. Twenty-five percent of patients also have neurological manifestations (e.g., microcephaly, sensorineural hearing loss, diminished or absent deep tendon stretch reflexes, cognitive impairment, and ataxia) [1].
Treatment for XP is supportive and depends on the affected organs, where skin lesions can be treated with freezing, topical agents (e.g., imiquimod and fluorouracil), oral agents (e.g., isotretinoin), or surgery for high-risk cases; eye neoplasms are managed surgically; and hearing loss can be improved with hearing aids. Preventive measures include avoiding UV exposure, which is aided by monitoring environments with UV light meters. Regular surveillance involves skin, eye, neurological, and hearing exams. Their life expectancy is 29 years in those with neurodegeneration and 37 years in those without [1].
Hemophagocytic lymphohistiocytosis (HLH) is a rare, frequently fatal hypersensitivity syndrome characterized by excessive activation and impaired downregulation of T-lymphocytes and macrophages, which leads to excessive proinflammatory cytokines release, destruction of blood cells, and tissue and organ damage [3]. It is estimated that HLH incidence is 1-2 per 1 million of the general population annually [4]. However, HLH is more common in the pediatric population, with an incidence of one per 50,000 children [5]. Diagnosis is based on HLH-2004 criteria [6].
HLH can be divided into primary (familial) HLH and secondary HLH. Primary HLH typically appears in early childhood and is often caused by mutations that affect cytotoxic cell function. Meanwhile, secondary HLH arises at any age due to external triggers that cause immune vulnerability, such as autoimmune diseases, malignancies, and infections. EBV is the most common infectious cause [7]. Other causes of HLH include cytomegalovirus, parvovirus B19, human immunodeficiency virus (HIV), and human herpesvirus-6 [8].
Treatment of HLH focuses on reducing hypercytokinemia and eliminating activated and infected cells. Treatment options for primary HLH include immunosuppressive, immunomodulatory, and cytostatic drugs; T-cell antibodies; agents targeting cytokines; and hematopoietic stem cell transplantation (HSCT) to suppress immunity [8]. Secondary HLH treatment aims to treat the underlying cause, along with the standard HLH treatment of chemotherapy and immunomodulatory agents [9]. G-CSF and blood products may be considered [8].
One rare cause of secondary HLH is Brucella bacteria, an intracellular infectious agent acquired by consuming unpasteurized dairy products, direct contact with infected animals, and inhalation. The bacteria can cause brucellosis, a chronic granulomatous disease requiring prolonged and combined antibiotic treatment [9].
As the most common zoonotic infection [10], Brucellosis usually presents with various constitutional symptoms (e.g., fever, malaise, and arthralgias), hepatomegaly, splenomegaly, and lymphadenopathy [9].
However, in rare cases, especially in regions with higher exposure to Brucella, it can lead to HLH [11].
2. Case Presentation
An 8-year-old Palestinian female patient, who was diagnosed with XP diagnosed within the first few months of life, presented with gross motor developmental delay and speech impairment. She began walking at 1.5 years of age and spoke her first two words by the age of one. She exhibits the typical cutaneous manifestations, including a diffuse skin rash with patches of discolored skin resembling severe aging, as shown in Figure 1. Her family history is notable for parental consanguinity and a brother with XP and intellectual disability.
In April 2024, the patient presented to a Jenin Governmental Hospital with a 3-day history of fever, generalized fatigue, abdominal pain, and arthralgia. Initial labs revealed pancytopenia, and Brucella serology was positive. She was hospitalized for 10 days. However, due to a lack of documentation in her medical records, the specific treatment plan administered during that admission is unknown. She was subsequently referred to An-Najah National University Hospital (NNUH) for bone marrow biopsy evaluation, which showed normocellular trilineage hematopoiesis with evidence of hemophagocytosis. She was discharged on a 6-week course of oral doxycycline and rifampin.
In July of 2024, the patient experienced recurrent episodes of fevers spiking up to 38°C managed with antipyretics by the parents. As the episodes persisted, further evaluation was undertaken. On July 27^th,^ 2024, an abdominal ultrasound showed splenomegaly (15 cm) and a right inguinal lymph node enlargement. On the fifth of August, the patient presented with fever, arthralgia, back pain, decreased appetite, and hypoactivity. Brucella testing was again positive. Laboratory results (Table 1) demonstrated persistent pancytopenia, elevated inflammatory markers (C-reactive protein and erythrocyte sedimentation rate), hyperferritinemia, and elevated triglycerides and lactate dehydrogenase (LDH), raising concerns for HLH. This prompted a second bone marrow biopsy.
Bone marrow aspirate findings confirmed the presence of hemophagocytes (Figure 2), supporting the HLH diagnosis. The patient was treated with rifampin (400 mg once daily for 6 weeks), doxycycline (50 mg twice daily for 6 weeks), and piperacillin/tazobactam (2000 mg IV). She responded well to treatment, as evidenced by normalization of her lab results and improvement in her symptoms.
3. Discussion
Brucellosis-induced HLH is rare, with limited cases reported in the literature. We conducted a comprehensive literature review using PubMed, Google Scholar, and other relevant databases but were unable to identify any previously reported cases of brucellosis-induced HLH in patients with XP. To the best of our knowledge, this is the first reported case. Our PubMed search using the terms “Brucella OR brucellosis” AND “HLH OR hemophagocytic lymphohistiocytosis” identified 18 case reports published between 2006 and 2024, describing 21 patients [12].
A prior literature review of 22 patients reported fever in all cases, splenomegaly in 7, hemoglobin levels below 9 g/dL in 7, neutrophil counts under 1 × 10^3^/μL in 13, platelet counts below 100 × 10^9^/L in 20, triglycerides above 265 mg/dL in 9, ferritin ≥ 500 ng/mL in 15, and fibrinogen levels under 150 mg/dL in 7. Blood cultures were positive in 14 patients, and bone marrow examination revealed hemophagocytosis in 16. These laboratory findings are consistent with our case, except for the platelet count (108 × 10^9^/L) and fibrinogen level (365 mg/dL). Soluble IL-2 receptor levels and NK cell activity were not investigated in most of the patients in the reviewed literature, nor in our patient.
Seventeen of the reviewed cases were treated with antibiotics alone, with complete recovery in 16, with a similar outcome to our patients. It is worth noting that in some reports, it was unclear whether certain laboratory tests had been performed. Comparison between our case and the published cases indicates strong parallels in both treatment approaches and outcomes [13]. This suggests that in infection-driven HLH, pathogen eradication can be sufficient, though immunosuppressive therapy may be necessary in severe or refractory cases [13].
The coexistence of these conditions carries important clinical implications. In XP, ultraviolet (UV) exposure triggers immune overactivation with elevated levels of interleukin-1 (IL-1), interleukin-6 (IL-6), and interferon-gamma (IFN-γ)—cytokines that also play central roles in HLH pathogenesis. This shared cytokine profile, with the addition of the fact that Brucella disrupts immune regulation via interleukin-10 (IL-10), a cytokine with potent anti-inflammatory effects. For this reason, the condition often requires the use of both antibiotics and immunosuppressants to prevent deterioration [8]. These immunopathological mechanisms also help explain why patients with XP are more predisposed to exaggerated inflammatory responses, thereby heightening the risk of HLH during infectious episodes.
Brucella's ability to persist within macrophages prolongs immune activation, driving cytokine overproduction (IFN-γ, TNF-α, and IL-6) and creating a “cytokine storm.” Immune dysregulation in XP further exacerbates this process. Defective macrophage bactericidal function, impaired dendritic cell antigen presentation, diminished NK cell cytotoxicity, and compromised T-cell responses collectively reduce infection control and prolong inflammation. This immune deficit likely contributed to our patient's recurrent fevers and prolonged hyperinflammatory state [14, 15].
Understanding the interaction between XP-related immune dysfunction and intracellular pathogens such as Brucella is essential. It suggests that XP patients in endemic regions may warrant heightened surveillance for HLH when presenting with systemic infections, and that clinicians should consider Brucella testing early in the diagnostic process.
4. Conclusion
This case represents the first reported case of brucellosis-induced HLH in a patient with XP. As a singular report, the findings cannot be generalized; the overlap in immune dysregulation between XP and HLH suggests a potential predisposition to severe infection-related hyperinflammation. Further studies and case series are needed to elucidate the relationship between XP, HLH, and brucellosis. Clinicians should maintain a high index of suspicion for HLH in XP patients presenting with systemic infection, particularly in Brucella-endemic areas. Long-term follow-up of such cases would provide critical insights into disease progression and treatment outcomes, particularly in XP patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Leung A. K. C. Barankin B. Lam J. M. Leong K. F. Hon K. L. Xeroderma Pigmentosum: An Updated Review Drugs in Context 20221111710.7573/dic.2022-2-5PMC 904548135520754 · doi ↗ · pubmed ↗
- 2Wolska H. Xeroderma Pigmentosum Przeglad Dermatologiczny 2006935555564
- 3Fisman D. N. Hemophagocytic Syndromes and Infection Emerging Infectious Diseases 20006660160810.3201/eid 0606.0006082-s 2.0-003448661811076718 PMC 2640913 · doi ↗ · pubmed ↗
- 4Cleves D. Lotero V. Medina D. Pediatric Hemophagocytic Lymphohistiocytosis: A Rarely Diagnosed Entity in a Developing Country BMC Pediatrics 2021211411418 https://bmcpediatr.biomedcentral.com/articles/10.1186/s 12887-021-02879-7 10.1186/s 12887-021-02879-734537050 PMC 8449481 · doi ↗ · pubmed ↗
- 5Hemophagocytic Lymphohistiocytosis (HLH)Children’s Hospital of Philadelphia https://www.chop.edu/conditions-diseases/hemophagocytic-lymphohistiocytosis-hlh%23;types
- 6Henter J. I. Horne A. C. AricóM. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis Pediatric Blood and Cancer 200748212413110.1002/pbc.210392-s 2.0-3384561913716937360 · doi ↗ · pubmed ↗
- 7Wu T. Tang L. Hu Y. Hemophagocytic Lymphohistiocytosis: Pathogenesis and Diagnosis Journal of Clinical Hematology. 2024379677682
- 8George M. R. Hemophagocytic Lymphohistiocytosis: Review of Etiologies and Management Journal of Blood Medicine 201456986 https://www.dovepress.com/nbsphemophagocytic-lymphohistiocytosis-review-of-etiologies-and-manage-peer-reviewed-fulltext-article-JBM 10.2147/jbm.s 4625524966707 PMC 4062561 · doi ↗ · pubmed ↗