Multigene Typing of Croatian ‘Candidatus Phytoplasma Mali’ Strains
Ivana Križanac, Martina Šeruga Musić, Jelena Plavec, Dijana Škorić

TL;DR
This study explores the genetic diversity of a plant pathogen in Croatia, revealing new genotypes and proposing a foundational sequence type for future research.
Contribution
The study introduces a multigene typing approach and novel primers for expanded phytoplasma sequence typing in Croatia.
Findings
Twenty different sequence types were identified in Croatian ‘Ca. P. mali’ strains.
A founder sequence type ST1 (A13–P10–S12–I21) was proposed based on the analysis.
Genetic diversity was found to be limited to two locations in north-western Croatia.
Abstract
Phytoplasmas (‘Candidatus Phytoplasma’) are intracellular pleomorphic plant pathogens belonging to the class Mollicutes. They colonize both plant hosts and insect vectors in their life cycle. Apple proliferation (AP) is one of the most important phytoplasmoses present in Europe, causing significant economic losses in apple production. The causal agent, ‘Ca. P. mali’, was identified in apple and Cacopsylla picta samples using both real-time PCR and nested PCR based on the amplification of 16S rDNA. The objective of this study was to gain deeper insights into the epidemiology of apple proliferation in Croatia. Variability of genetic markers other than 16S rRNA was used for characterization of strains. Four molecular markers differing in level of conservation, aceF, pnp, imp, and secY, were selected in line with previously typed fruit tree phytoplasmas. New genotypes were discerned for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
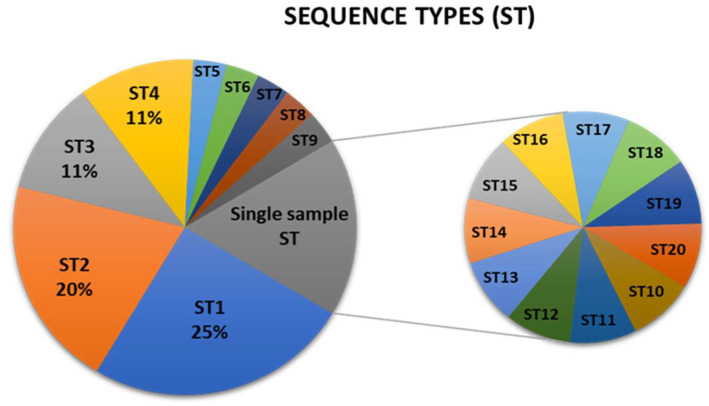 Figure 1
Figure 1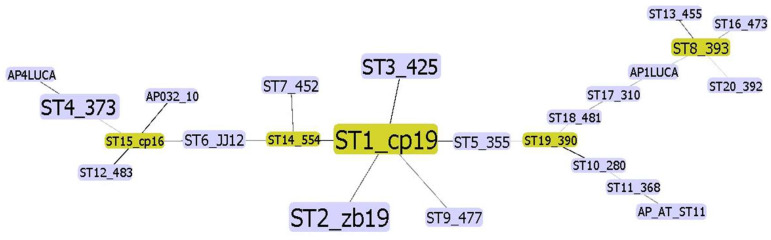 Figure 2
Figure 2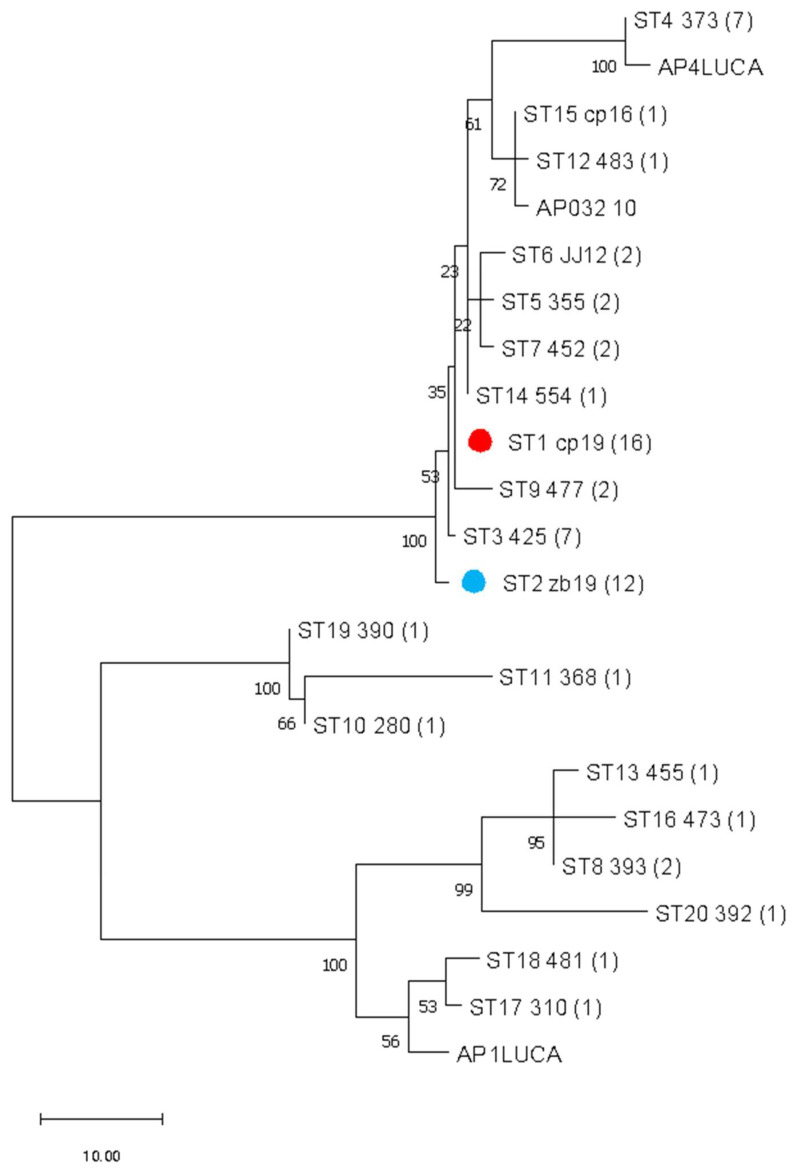 Figure 3
Figure 3- —Croatian Ministry of Agriculture
- —University of Zagreb
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytoplasmas and Hemiptera pathogens · Plant Pathogenic Bacteria Studies · Cocoa and Sweet Potato Agronomy
1. Introduction
Phytoplasmas (‘Candidatus Phytoplasma’) are wall-less prokaryotes from the class Mollicutes that induce diseases in more than a thousand plant species worldwide [1]. They are limited to the phloem conducting elements in host plants, while in insect vectors they can be found in various organs and tissues, including the hemolymph and reproductive organs [1,2]. Their phylogeny is mainly based on the common bacterial phylogenetic marker 16S rRNA gene [3]. Phytoplasma genomes are highly reduced and are among the smallest bacterial genomes described to date, regardless of their adaptation to both plant and insect hosts. Inability to cultivate phytoplasmas in axenic culture still hinders their detailed characterization as well as the acquisition of high-quality genomic DNA. Despite many difficulties, more than 50 draft or complete genomes have been sequenced in the last 20 years [4,5], with the first one being the OY-M strain of the subgroup 16SrI-B [6].
Fruit tree phytoplasmas occurring in Europe, ‘Ca. P. mali’, ‘Ca. P. pyri’ and ‘Ca. P. prunorum’, belong to the same phylogenetic cluster [7]. Their chromosomes are linear, which is an unusual feature for phytoplasmas and bacteria in general [8]. Multi-locus sequence typing (MLST) was proposed as a method applicable for characterization of many pathogenic bacteria [9]. This approach was used to differentiate phytoplasma strains and enhance understanding of the molecular epidemiology of European fruit tree phytoplasmas belonging to the ribosomal group 16SrX [10].
Characteristic symptoms of apple proliferation (AP) include the formation of “witches’ brooms”, enlarged stipules, and reduced fruit size with elongated pedicels [11]. The described symptoms were observed in apple orchards in north-western Croatia more than 40 years ago, and pleomorphic mycoplasma-like cells were detected in phloem tissues [12]. Although surveys of both ‘Ca. P. mali’ and ‘Ca. P. pyri’ presence in Croatian orchards started in 2003, ‘Ca. P. mali’, the causal agent of AP, was not confirmed in apple tree samples until 2011 [13,14]. Two psyllid species, Cacopsylla picta (Förster) [15] and C. melanoneura (Förster) [16], are identified as vectors of ‘Ca. P. mali’. Population dynamics of psyllids were monitored from 2005 to 2007, and both C. melanoneura and C. picta were found to be present and widespread [17]. With the availability of ‘Ca. P. mali’ genome [8], this study was designed to optimize the MLST scheme, enabling more accurate and robust molecular characterization of this phytoplasma. In parallel, it aimed to improve the understanding of apple proliferation epidemiology in Croatian apple orchards.
2. Materials and Methods
2.1. Plant and Insect Samples
Samples infected with ‘Ca. P. mali’ from a previous study, one C. picta and 60 apple tree samples, were used for MLST [14]. In 2016, an additional 176 C. picta, 34 C. melanoneura, and 12 apple tree samples were collected mainly in the western continental part of Croatia, where the AP disease pressure was the highest, with a particular focus on psyllid monitoring. Samples were tested for ‘Ca. P. mali’ using both real-time PCR [18] and nested PCR/RFLP methods [19,20,21] as previously described [14]. A total of 423 samples were tested for phytoplasma presence, of which 74 positive samples were used for MLST (Table S1).
2.2. MLST Primers and PCR Conditions
Primers designed to amplify the entire aceF gene were used for both direct PCR and sequencing, as previously described [14]. New primers were designed based on the available AP phytoplasma genome [8] to amplify fragments longer than complete coding regions of imp, secY, and pnp genes. Due to the amplicon size, additional primer pairs were needed for both nested PCR and sequencing for pnp and secY genes (Table 1). In a nested PCR reaction, 1 µL of direct PCR mix was used as a template. Both direct and nested PCR conditions for aceF, pnp, and secY amplification were the same: initial denaturation at 94 °C for 4 min, followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at 52 °C for 2 min, extension at 68 °C for 3 min, and a final extension step of 7 min at 68 °C. To amplify the imp fragment in a direct PCR, the annealing and extension temperatures were 51 °C and 66 °C, respectively, with the same duration and number of cycles. Amplicons were custom sequenced (Macrogen Europe, Amsterdam, The Netherlands) on both strands for aceF and imp. For secY, additional internal primers were used for sequencing to increase coverage due to the amplicon size and to increase the coverage of the most variable region of the secY gene (Table 1).
2.3. Sequence Analysis
Raw nucleotide sequences were assembled and edited using both the Sequencher^®^ 4.7 software (Gene Codes Corporation, Ann Arbor, MI, USA, http://www.genecodes.com/) and Geneious 10.1.3 ([22], http://www.geneious.com), and aligned with ClustalX 2.0 [23]. A nucleotide BLAST search was performed on the NCBI website (https://blast.ncbi.nlm.nih.gov/Blast.cgi) against the GenBank core nucleotide database using the Megablast option optimized for highly similar sequences with default algorithm parameters. Consensus sequences of all four genes for each sample were concatenated using Geneious 10.1.3 [22] and analyzed using PhyloViz 2.0 [24]. Phylogenetic analyses were performed with MEGA 11 [25] using the maximum parsimony method with a bootstrap test (500 replicates) to support the inferred clades [26]. As amplicons and sequenced fragments exceeded the coding region for all four genes, both ExPasy [27] (http://web.expasy.org/translate/) and MEGA 11 [25] were used to identify open reading frames for each consensus sequence. Obtained sequences were trimmed in silico in two ways: first, to the length of reference sequences for each genotype of ‘Ca. P. mali’ [10] to enable comparison and continuation of the genotype labeling system, and second, to the length of the coding region to verify the accuracy of the consensus sequence. For each sample, a combination of aceF, pnp, imp, and secY genotypes was used to form a sequence type (ST), with the assigned ST number in the order of their prevalence.
3. Results
In a five-year period (2011–2014 and 2016), a total of 204 apple trees and 219 psyllid samples were analyzed using both nested PCR/RFLP and real-time PCR methods. Along with the previously published 61 positive samples [14], two apple and 11 C. picta samples were found to be infected with ‘Ca. P. mali’ in additional monitoring in 2016 (Table S1). None of the tested C. melanoneura harbored the phytoplasma. Out of 74 positive samples that were included in MLST, all four genetic markers were successfully typed for 64 positive samples (Table S2, Figure S1).
3.1. aceF, pnp, imp, and secY Genotyping
For the aceF fragment of approximately 790 bp in length, five of the eight previously described genotypes [10,28] were identified in Croatian samples, along with a new one labeled A27 (Table S2 and Figure S2). The predominant genotype, A13, was present in 67% of the analyzed samples. The unrooted phylogenetic tree obtained through the analysis of the full-length (1260 bp) aceF shows strong support for A27 genotype divergence (Figure S8).
Analysis of a 512 bp pnp gene fragment revealed seven distinct genotypes (Figure S3). Of the five previously described genotypes [10], only P9 and P10 are present in our samples, with P10 being the prevalent one in almost 70% of the samples (Table S2). Two new genotypes were identified, P17 and P18, with P18 found in only one apple sample (Figure S3 and Table S2). An unrooted phylogenetic tree based on the analysis of the complete pnp gene expectedly showed greater diversity because of the more than four times longer sequence (Figure S9).
Out of ten already described imp genotypes [10,28], five were found in Croatia together with six new ones (I36–I41). It is expectedly the most variable of the four typed genes (Figures S4 and S5, Table S2). The prevalent genotype, I21, was found in almost 50% of the samples (Table S2). A BLAST search of the newly identified genotype sequences against the GenBank nucleotide database revealed imp gene sequences identical to our new genotypes (Table S4). However, these entries had not been annotated and labeled according to the MLST scheme used in this study [29].
Analysis of nucleotide sequences of the secY gene fragment resulted in a phylogenetic tree separating into 12 genotypes (Figure S6). In addition to 7 known [10,28], 5 new genotypes (S17–S21) were identified in the samples from this study (Table S2). The genotype S12 was dominant and found in over 67% of the samples. Within the variable region of the secY gene, compared to the reference sequence of the strain AT ‘Ca. P. mali’ with a length of 1245 bp, there was either an insertion of three nucleotides (S17, sample 392) or deletions of multiple nucleotides (genotypes S10 and S12). As a result, the coding region length varies from 1227 bp (S11, S12, S19, and S18) to 1248 bp (S17). The secY gene fragment covers the most variable region (positions 397–438/1248 bp, Figure S7), and only a small number of significant nucleotide changes are found outside this section. Therefore, the unrooted phylogenetic tree based on the 1227–1248 bp secY gene sequence largely supports the tree obtained from the phylogenetic analysis of the shorter fragment (Figure S10).
Erroneously, labels I31 and S13 that had already been used for ‘Ca. P. pyri’ imp and secY genotypes from Azerbaijan [10] were used for the new genotypes in a more recent study [28]. Therefore, we propose correcting the genotype labels to avoid ambiguity, starting with the first available I35 and S16, respectively. These proposed labels are used throughout figures and tables in this study (Tables S2 and S4, Figures S4 and S6).
3.2. Sequence Types (STs) and Analysis of Concatenated Sequences
The obtained profile of genotype combinations for each sample was grouped to a sequence type (ST) and labeled in order of their frequency (Table S2). There are 20 different sequence types, with ST1 (A13–P10–I23–S12) represented in 25% of the Croatian samples (Figure 1). The ST4 (A15-P17-I21-S10) contains the new genotype P17 and is present in more than 10% of the samples. The ST2 profile (A13–P10–I21–S12) is the most common in C. picta, present in 50% of the insect samples (Figure 1, Table S2). Eleven out of 20 STs are represented in a single sample, mainly due to imp and secY variability, although ST11 and ST14 harbor known genotypes that are not frequent in Croatia (Figure 1, Table S2).
Concatenating all four gene sequences allowed simultaneous and comprehensive analysis of the ‘Ca. P. mali’ variability targeted in MLST. Analysis of concatenated sequences resulted in a finer resolution because all nucleotide differences are considered. In addition to interconnectedness shown in the phylogenetic network, the number of strains of the same profile is also considered and graphically represented by the node size (Figure 2). The frequency of each profile is used to select the founder ST for each cluster. Therefore, ST1 is considered an overall founder sequence type, and five clusters are suggested by analyzing this dataset (Figure 2). The unrooted phylogenetic tree inferred using the maximum parsimony analysis of concatenated aceF, pnp, imp, and secY sequences (Figure 3) largely supports the results represented in the network (Figure 2), with the presence of four main clusters.
4. Discussion
Following the results of a systematic survey of apple orchards, efforts were made to characterize Croatian ‘Ca. P. mali’ strains in more detail in order to obtain a better insight into the epidemiology of this important plant pathogen. However, since the trees exhibiting symptoms reminiscent of apple proliferation were preferentially sampled, the percentage of infected apple trees in this study does not necessarily represent the overall apple infection rate in the country. The frequency of naturally infected psyllids, C. picta and C. melanoneura, as well as their ability to transmit the disease, differs significantly in countries and regions where the disease is present and well-studied [16,30]. In Croatia, C. melanoneura populations are more numerous than C. picta, and yet C. picta adults are present in apple orchards for a longer period before migration to overwintering coniferous hosts. This possibly increases their potential for phytoplasma acquisition [17]. Transovarial transmission of the phytoplasma to offspring has been proven for C. picta [31], thus bypassing the process of acquisition from the infected plant and multiplication in the vector. With more than 200 psyllid samples tested, we can positively conclude that the major AP phytoplasma vector in Croatia is C. picta. This vector infection pattern is consistent with research results from Germany, Switzerland, and France [32].
Primers developed in this study (Table 1) successfully amplified the target regions of all four MLST genes in the majority of samples. Novel genotypes were identified for each of the four genes analyzed. For aceF, genotype A13 was predominant (Figure S2 and Table S2), aligning with previous reports from strains collected in France, Italy, and Germany [10]. In ‘Ca. P. prunorum’, aceF genotypes A6 and A8 [10] have been associated with hypovirulent phytoplasma strains [33]. However, due to the lack of data on the biological properties of the Croatian ‘Ca. P. mali’ strains, it is currently not possible to infer similar associations from our findings. Phylogenetic analysis of the pnp gene fragment revealed two new genotypes—P17 and P18 (Figure S3 and Table S2). Genotype P17 was fairly frequent and is present in eight samples, while P18 was recorded in only one apple tree sample. The prevalent genotype P10 was present in almost 70% of the samples. From the results of genotyping European strains, the dominant genotype was P11 [10], which is not present in Croatia. When the entire coding region was analyzed, samples that were grouped within genotypes P9 and P10 separated into distinct branches (Figure S9), which is not surprising given that the complete coding region is four times longer than the fragment usually used in genotyping [10]. The imp gene was expectedly the most variable of the four typed genes (Figure S4, Table S2). This gene encodes the immunodominant membrane protein that is located on the cell surface and is thus exposed to positive selective pressure. Due to its role in interactions with both the apple and the insect vector, it is relevant in the molecular pathology of the disease [34,35]. Six novel genotypes (I36–I41) were identified in Croatia (Figure S4 and Table S2). This variability is common and consistent with studies of immunodominant membrane proteins from other phytoplasma species [35,36,37,38]. As many as 87 out of approximately 507 nucleotide positions in the imp gene sequence are variable, with the most significant difference being deletions within the sequence, causing the length to vary from 498 to 507 bp (Figure S5). The dominant genotype is I21, the same as at the European level [10,28]. The three genotypes identified in the largest number of samples (I21, I22, and I23) were grouped in the same cluster (Figure S4). Interestingly, all new genotypes were found exclusively in apple samples from two locations less than 5 km apart (Figure S1 and Table S2), while in C. picta, only I21 and I23 genotypes were present (Table S2). Changes in the imp gene in apple samples probably bring advantages, such as evading the host immune response [34]. In C. picta, phytoplasma must pass through various tissues and multiply to a sufficient concentration for successful transmission from salivary glands to the host [35,39,40]. This suggests that these two genotypes might be optimal for this translocation. The secY gene, which encodes the central subunit of the membrane transport system, has long been used as an additional phylogenetic marker for better differentiation of closely related phytoplasmas within the same 16S rRNA group [41]. Phylogenetic analyses of the obtained sequences show higher than expected variability (Figures S6 and S7, Table S2). Such variability of the secY gene is not straightforward and was not expected for this housekeeping gene, especially since MLST of the European fruit tree phytoplasma strains had shown that this gene is less variable than aceF and pnp [10]. The previously described genotype S12, which was dominant in ‘Ca. P. mali’ strains from five European countries [10], is present in by far the largest number of Croatian samples too (almost 70%) (Figure 1, Table S2). In this study, five new genotypes (S17–S21) were described (Figure S6 and Table S2). The same as with the imp gene, all new genotypes are present exclusively in apple samples from two locations in north-western part of Croatia (Figure S1 and Table S2). Introducing and naming new genotypes does not come without difficulties and we encountered inadvertently introduced erroneous labels for newly recorded genotypes for both imp and secY [28]. To ensure labeling accuracy moving forward, we propose reassigning the labels to the first available ones, namely I35 and S16, respectively.
Results of MLST revealed new genotypes and great diversity in two close-by locations in the north-western part of Croatia, Donji Mihaljevec and Sveta Marija (Figure S1 and Table S2). Apples have been traditionally grown in this region for a long time, and there are records indicating that ‘Ca. P. mali’ has been present here at least since the 1980s, when symptoms were first observed [12]. This long-lasting coevolution of the host and pathogen is one possible explanation for this diversification. Interestingly, this diversity is not represented in C. picta samples that harbor only one new genotype, P17 (Table S2). Results of genotyping individual genes were combined to form 20 different sequence types (Figure 1, Table S2). ST1 (A13–P10–I23–S12) is the most frequent one and is represented in 25% of the Croatian samples (Figure 1, Table S2). The profile ST2 (A13–P10–I21–S12) is the most common in C. picta, present in more than 50% of the insect samples (Figure 1, Table S2). Two sequence types differ only in two imp genotypes (I23 vs. I21). Since ST2 is dominant in C. picta samples, it can be hypothesized that I21 is significant in vector colonization and transmission. This is consistent with findings in Slovenia, where only the novel genotype I35 was found in the C. melanoneura sample [28]. Ten out of 20 STs are represented in a single sample, mainly due to imp and secY variability (Figure 1, Table S2).
Analysis of concatenated sequences suggested that the most frequent ST1 can be considered as the founder sequence type (Figure 1, Figure 2 and Figure 3). The unrooted phylogenetic tree inferred using the maximum parsimony analysis of concatenated aceF, pnp, imp, and secY sequences (Figure 3) largely supports the results represented in the network (Figure 2), with four main clusters. With only 18 previously fully genotyped strains from Austria, Switzerland, Germany, France, Italy, and Romania [10], 35 from neighboring Slovenia, and 64 as a part of this study, the dataset was still insufficient to create clear conclusions on the geographic distribution, origin, and virulence of individual strains. Nevertheless, the results of this study contribute to understanding the prevalence of individual genotypes in relation to the plant or insect host.
To improve and expand genotyping for phytoplasmas, it would be essential to agree on an MLST system for genes that have proven to be very informative, like hflB [42], SAP11 [43], or ribosomal protein (rp) genes [44]. The genome of ‘Ca. P. mali’ has been fully sequenced and annotated [8], enabling the search for additional, epidemiologically relevant, and evolutionary interesting genes. Furthermore, the creation of a database with representative genotype sequences according to uniform criteria and protocols would allow for the structured comparison of an increasing number of strains from across Europe. This would advance future applied research, such as the selection of apple cultivars resistant or tolerant to apple proliferation disease. The use of specific primers that allow amplification of the entire coding region for all four genes provided deeper insights into molecular characterization and may serve as a foundation for further in silico research of protein structure. Genotyping results are useful for understanding the epidemiology of phytoplasmas, and this study is a step toward creating a standardized MLST system for ‘Ca. P. mali’.
5. Conclusions
New genotypes are described in Croatian apple and C. picta samples for all four genes used in MLST: one for aceF, two for pnp, six for imp, and five for secY genes. All new imp and secY genotypes are found exclusively in apple samples. Twenty different sequence types (ST) are described, with 50% represented with only one sample, mainly due to imp and secY variability. Dominant ST1 was found in 25% of the samples, yet in C. picta samples, ST2 was present in more than half of the samples. Specific primers enabled amplification of the full coding region for all four genes, improving molecular characterization and supporting future in silico protein studies. The genotyping results provide valuable insights into the epidemiology of ‘Ca. P. mali’, with the dominance of ST1 highlighting its potential role in disease persistence and spread. This study contributes to the development of an expanded and improved MLST system for more precise characterization of phytoplasma strains.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Marcone C. Molecular Biology and Pathogenicity of Phytoplasmas Ann. Appl. Biol 201416519922110.1111/aab.12151 · doi ↗
- 2Lee I.-M. Davis R.E. Gundersen-Rindal D.E. Phytoplasma: Phytopathogenic Mollicutes Annu. Rev. Microbiol.20005422125510.1146/annurev.micro.54.1.22111018129 · doi ↗ · pubmed ↗
- 3IRPCM ‘Candidatus Phytoplasma’, a Taxon for the Wall-Less, Non-Helical Prokaryotes That Colonize Plant Phloem and Insects Int. J. Syst. Evol. Microbiol.2004541243125510.1099/ijs.0.02854-015280299 · doi ↗ · pubmed ↗
- 4Wei W. Zhao Y. Phytoplasma Taxonomy: Nomenclature, Classification, and Identification Biology 202211111910.3390/biology 1108111935892975 PMC 9394401 · doi ↗ · pubmed ↗
- 5Kirdat K. Tiwarekar B. Sathe S. Yadav A. From Sequences to Species: Charting the Phytoplasma Classification and Taxonomy in the Era of Taxogenomics Front. Microbiol.202314112378310.3389/fmicb.2023.112378336970684 PMC 10033645 · doi ↗ · pubmed ↗
- 6Oshima K. Kakizawa S. Nishigawa H. Jung H.-Y. Wei W. Suzuki S. Arashida R. Nakata D. Miyata S. Ugaki M. Reductive Evolution Suggested from the Complete Genome Sequence of a Plant-Pathogenic Phytoplasma Nat. Genet.200436272910.1038/ng 127714661021 · doi ↗ · pubmed ↗
- 7Seemüller E. Schneider B. ‘Candidatus Phytoplasma mali’, ‘Candidatus Phytoplasma pyri’ and ‘Candidatus Phytoplasma prunorum’, the Causal Agents of Apple Proliferation, Pear Decline and European Stone Fruit Yellows, respectively Int. J. Syst. Evol. Microbiol.2004541217122610.1099/ijs.0.02823-015280295 · doi ↗ · pubmed ↗
- 8Kube M. Schneider B. Kuhl H. Dandekar T. Heitmann K. Migdoll A.M. Reinhardt R. Seemüller E. The Linear Chromosome of the Plant-Pathogenic Mycoplasma “Candidatus Phytoplasma mali”BMC Genomics 2008930610.1186/1471-2164-9-30618582369 PMC 2459194 · doi ↗ · pubmed ↗