Intermedin Inhibits DNA Damage-Promoted Senescent Phenotype Transition of Vascular Smooth Muscle Cells in Aorta by Activating NAMPT/PARP1 in Mice
Deng-Ren Ji, Yao Chen, Han-Xu Zhu, Shi-Meng Liu, Ning Wu, Ya-Rong Zhang, Jie Zhao, Yan-Rong Yu, Mo-Zhi Jia, Ling Han, Chao-Shu Tang, Lei-Lei Chen, Ye-Bo Zhou, Yong-Fen Qi

TL;DR
This study shows that Intermedin helps prevent aging-related changes in blood vessel cells by reducing DNA damage through a specific pathway.
Contribution
The study reveals a novel mechanism by which Intermedin inhibits vascular smooth muscle cell senescence via the NAMPT/PARP1 pathway.
Findings
Intermedin reduces DNA damage markers in vascular smooth muscle cells.
Intermedin activates NAMPT and PARP1 to increase NAD+ levels and prevent cell aging.
Blocking NAMPT or PARP1 negates the protective effects of Intermedin.
Abstract
Background and aims: The senescent phenotype transition of vascular smooth muscle cells (VSMCs) is a crucial risk factor for the occurrence and development of vascular diseases. Intermedin (IMD) has various protective effects on cardiovascular diseases. In this study, we aimed to explore the role and the related mechanism of IMD in the senescent phenotype transition of VSMCs of aorta in mice. Methods: The senescent phenotype transition of VSMCs was induced by angiotensin II (Ang II) administered by mini-osmotic pumps in Adm2fl/fl and Adm2fl/flTagCre mice. Mouse VSMCs from aorta were used in in vitro experiments. Results: The aortic mRNA level of IMD, namely Adm2, was significantly decreased in Ang II-treated mice. Senescence-associated β-galactosidase activity and protein expressions of p16 and p21 were increased in the aortas of Adm2fl/flTagCre mice, which were further elevated in Ang…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
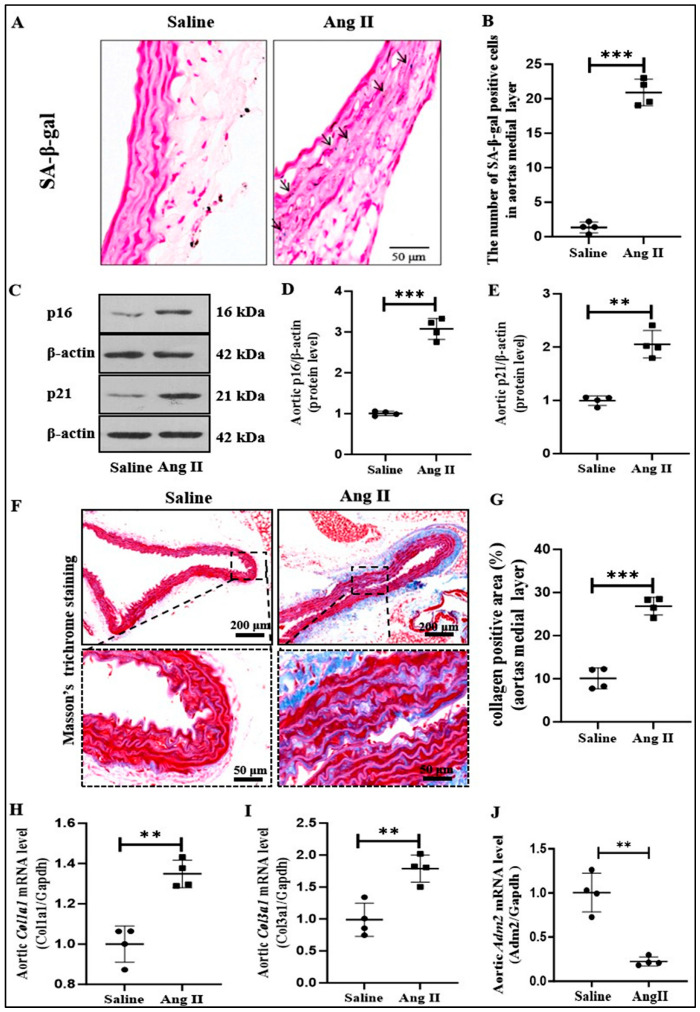 Figure 1
Figure 1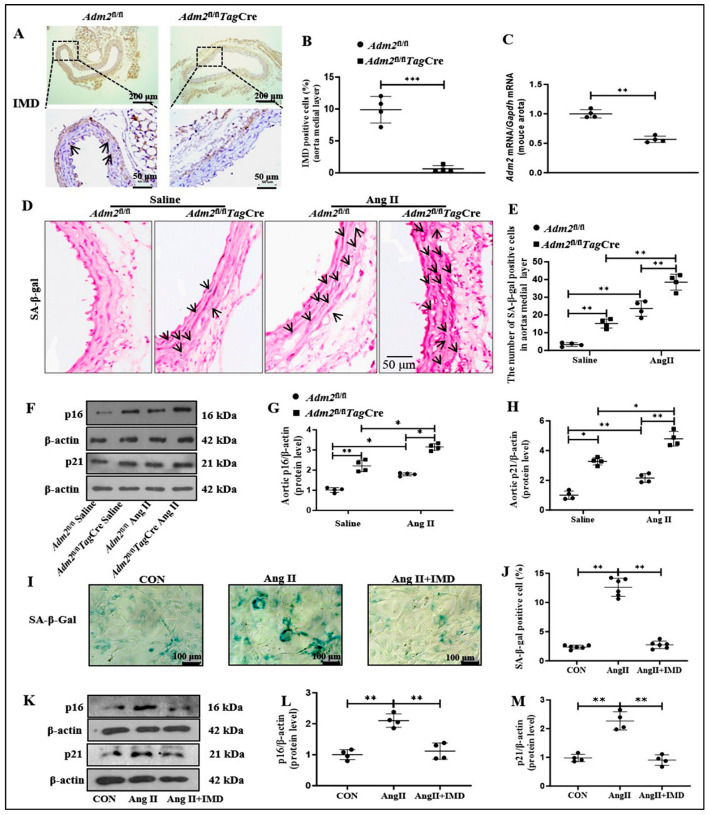 Figure 2
Figure 2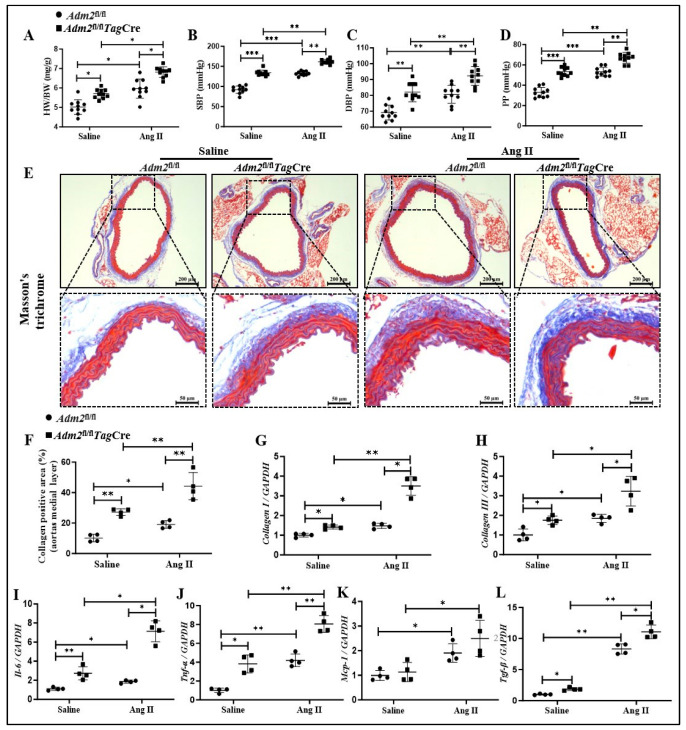 Figure 3
Figure 3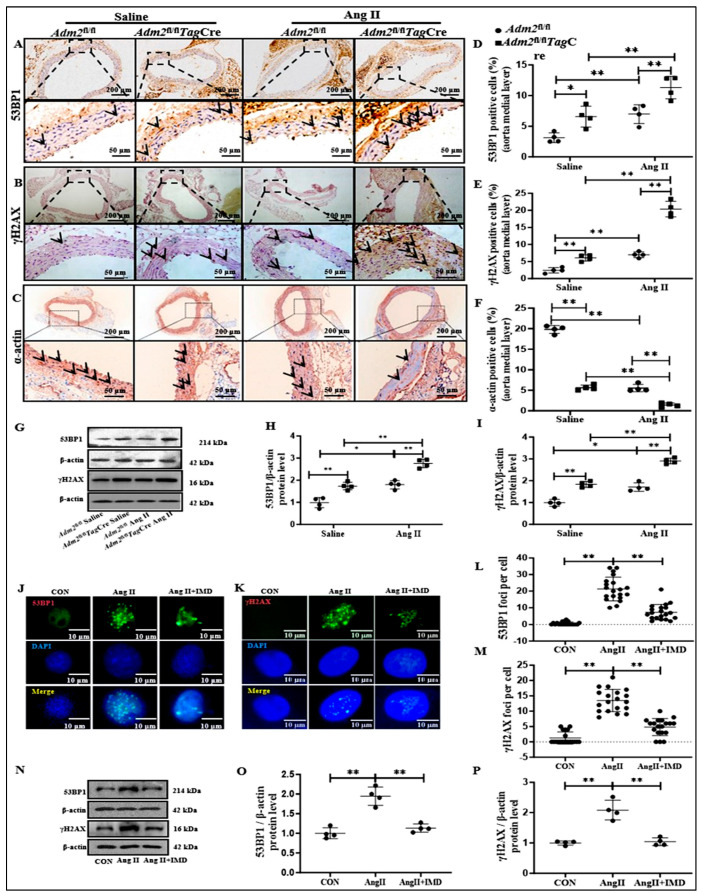 Figure 4
Figure 4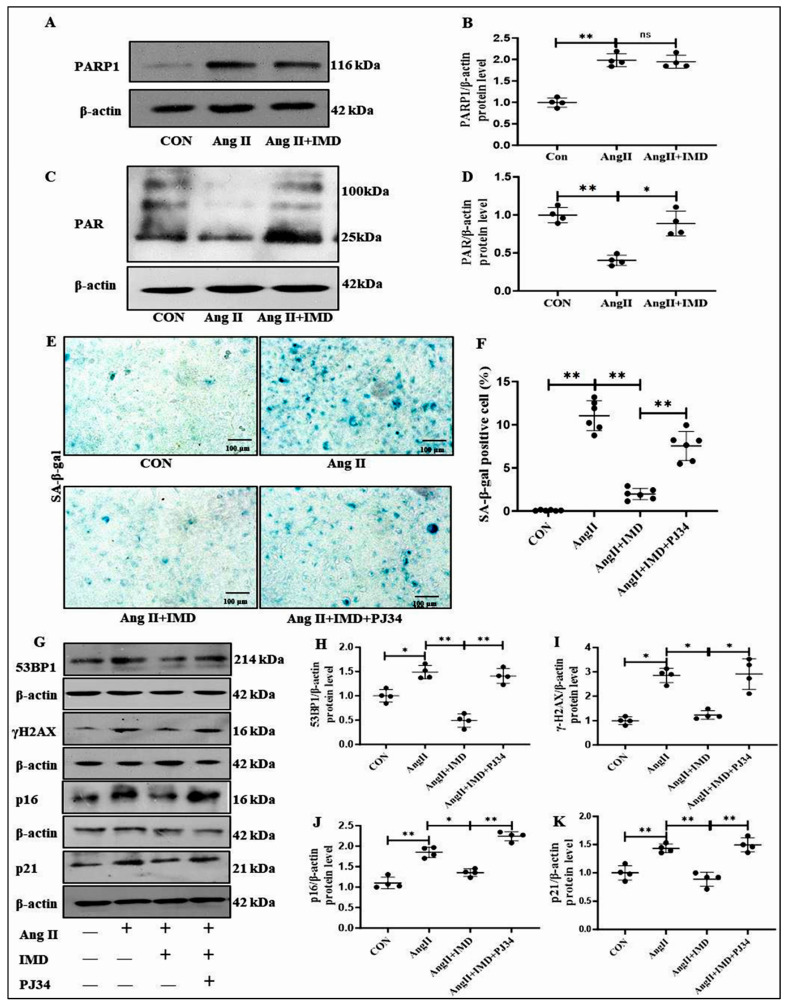 Figure 5
Figure 5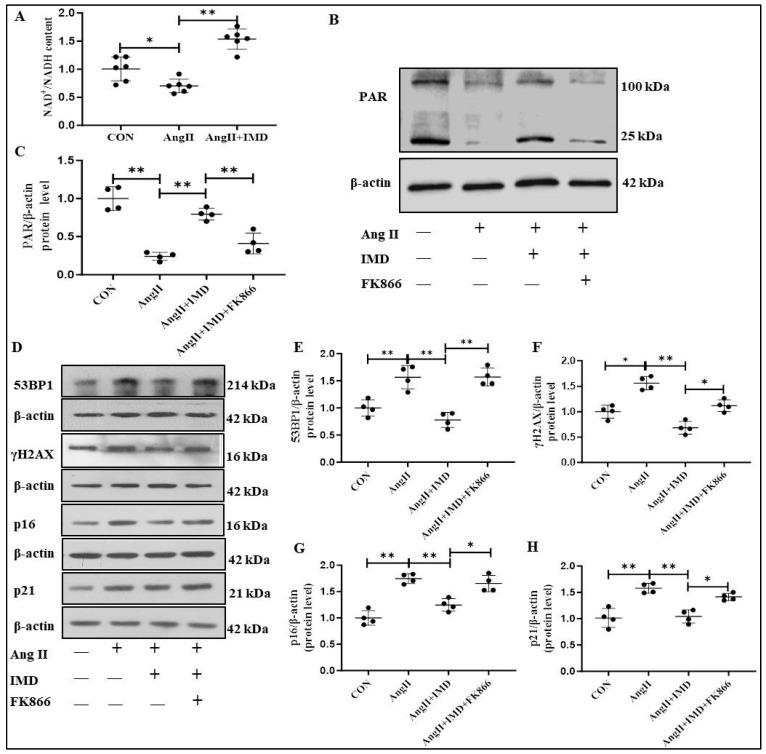 Figure 6
Figure 6- —the National Natural Science Foundation of China
- —the Beijing Natural Science Foundation
- —Health Commission of Jiangsu Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAtherosclerosis and Cardiovascular Diseases · Adipokines, Inflammation, and Metabolic Diseases · Chemokine receptors and signaling
1. Introduction
The aortic wall is composed of three structurally distinct layers: intima, media, and adventitia. The arterial media is usually the thickest layer; it provides support for the vessel and alters vessel diameter to regulate blood flow and blood pressure. Vascular smooth muscle cells (VSMCs) are the main cells of tunica media and play an important role in maintaining vascular function depending on its systolic phenotype [1]. Moreover, the abnormal VSMC’s plasticity and signaling are highly relevant to early vascular aging [2]. And increasing experimental data indicated that the senescent phenotype transition of VSMCs (from systolic phenotype to senescent phenotype) plays a pivotal role in inducing aging-related vascular remodeling [3,4]. In addition, a previous study found that attenuating angiotensin II (Ang II)-induced VSMCs senescence could effectively alleviate aging-associated vascular remodeling [5]. The VSMCs is an initial and fundamental aging process in which cells permanently withdraw from the cell cycle under the stimulation of a range of endogenous and exogenous stressors such as telomere dysfunction, DNA damage, reactive oxygen species (ROS), and paracrine signals and undergo distinctive phenotypic changes [6]. It is necessary to inhibit the senescent phenotype transition of VSMCs in the prevention and treatment of vascular aging and related diseases.
DNA damage and genome instability play a crucial role in the initiation and progression of cellular senescence [7]. DNA damage caused by endogenous and exogenous stimuli triggers the DNA damage response (DDR), which in turn affects cell fate, repair, or senescence [8]. To deal with DNA damage, a mature DNA repair system has evolved, which can restore the correct base sequence in most cases. However, disturbance of the DNA repair system may lead to sustained DNA damage [7]. In response to sustained DNA damage, cells undergo phenotypic transitions such as the generation of a bioactive secretome, namely the senescence-associated secretory phenotype, represented by the increased protein expressions, such as pro-inflammatory cytokines, chemokines, and growth factors [9]. The transcriptional activation of p16 and p21, two cyclin-dependent kinase inhibitors, is also induced by DDR and leads to cell cycle arrest [10]. Therefore, the accumulation of DNA damage over time is a fundamental driver of aging. Attenuation of DNA damage might delay the senescent phenotype transition of VSMCs and then reverse vascular aging.
Intermedin (IMD), also known as adrenomedullin 2 (ADM2), is a secreted peptide belonging to the calcitonin gene-related peptide (CGRP) superfamily [11,12]. The human IMD gene encodes a prepropeptide composed of 148 amino acids, which has a signal peptide at its N-terminus. By proteolytic cleavage at Arg93-Arg94, IMD_1-53_ can be produced from pre-pro-IMD [13]. IMD exerts its biological effects by combining with calcitonin receptor-like receptor (CRLR) and receptor activity modifying protein (RAMP)1/2/3 [11]. It plays an important role in maintaining cardiovascular homeostasis and exerting a potential beneficial role in cardiovascular diseases (CVDs). Increasing evidence shows that IMD has a protective effect on vascular abnormalities, including vascular calcification [14,15] and abdominal aortic aneurysm [16]. Moreover, our previous study found that IMD levels were significantly decreased in aortas of aging rat and senescent VSMCs, and supplementation with exogenous IMD significantly reduced the protein levels of p16 and p21 in aging rat aortas [17], suggesting that IMD might be involved in the regulation of senescent phenotype transition of VSMCs. In this study, we aimed to investigate the role of IMD in Ang II-induced senescent phenotype transition of VSMCs and the possible mechanism involving DNA damage.
2. Results
2.1. IMD Expression and Ang II-Induced Senescent Phenotype Transition of VSMCs in Aorta of Mice
In this study, mice with initial changes in senescent phenotype transition of VSMCs in aorta of mice were constructed by Ang II subcutaneous buried pump [17,18]. First, the changes in senescent markers in senescence-associated β-galactosidase (SA-β-gal) activity and protein levels of cyclin-dependent kinase inhibitors p16 and p21 in the aorta of mice were detected. Compared to controls, Ang II-induced membrane SA-β-gal activity was significantly higher in mouse aorta (Figure 1A,B), and protein levels of p16 and p21 were also significantly increased (Figure 1C–E). Vascular fibrosis is one of the features of vascular aging [19], and the area of Ang II-induced aortic fibrosis was significantly increased in mice compared with controls (Figure 1F,G). Real-time PCR results also showed that the aortic mRNA levels of Col1a1 and Col3a1 were significantly higher in Ang II-treated mice than in the control group (Figure 1H,I). In conclusion, Ang II-induced senescent phenotype transition of VSMCs in aorta as shown by the increased activity of aging-related markers SA-β-gal, protein levels of p16 and p21 proteins, and fibrosis in aorta of mice, indicated that the senescent phenotype transition of VSMCs in aorta was successfully constructed. Moreover, the mRNA level of IMD, namely Adm2, in the aortic tunic media of the Ang II-treated group was significantly decreased when compared with the control group (Figure 1J), suggesting that IMD may be involved in the pathogenesis of aortic aging induced by Ang II.
2.2. Deficiency in Endogenous IMD Exacerbated Ang II-Induced Senescent Phenotype Transition of VSMCs in Aorta
To clarify the role of endogenous IMD in senescent phenotype transition of VSMCs in aorta, we evaluated whether SMCs-specific ablation of IMD would exacerbate Ang II-induced senescent phenotype transition of VSMCs in the aorta of mice. Immunohistochemistry staining verified that IMD was deficient in the aortic media layer of Adm2^fl/fl^TagCre mice (Figure 2A,B). Moreover, the mRNA level of Adm2 in the aorta of Adm2^fl/fl^TagCre mice was significantly lower than that in the Adm2^fl/fl^ group (Figure 2C). Compared with Adm2^fl/fl^ Saline mouse aortas, SA-β-gal activity was markedly upregulated in the aortic media layer of Adm2^fl/fl^ mice treated with Ang II and further increased in Adm2^fl/fl^TagCre mice treated with Ang II (Figure 2D,E). In addition, Ang II treatment strikingly elevated the protein levels of p16 and p21 in the aorta of Adm2^fl/fl^TagCre mice (Figure 2F–H). These results suggested that IMD deficiency may exacerbate Ang II-induced senescent phenotype transition of VSMCs in aorta. More importantly, SA-β-gal activity and the protein levels of p16 and p21 were also increased in the aorta of the Adm2fl/flTagCre mice compared to the Adm2fl/fl mice. These results suggest that IMD deficiency in VSMCs may exacerbate senescent phenotype transition and further promoted Ang II-induced senescent phenotype transition of VSMCs in aorta.
Next, we applied exogenous IMD_1-53_ to observe the effect of IMD on the senescent phenotype transition of VSMCs in vitro. SA-β-gal staining showed that Ang II treatment significantly increased SA-β-gal activity in VSMCs isolated from Adm2^fl/fl^ mice, while exogenous IMD_1-53_ significantly decreased SA-β-gal activity induced by Ang II (Figure 2I,J). In addition, the protein levels of p16 and p21 were significantly increased in Ang II-treated VSMCs compared to the control VSMCs, which was reversed by IMD_1-53_ supplementation (Figure 2K–M). Taken together, these results suggested that IMD could also inhibit Ang II-induced senescent phenotype transition of VSMCs in vitro.
2.3. Deficiency in Endogenous IMD Exacerbated Ang II-Induced Vascular Fibrosis and Prosenescent Factors Expression
Given that collagen synthesis is upregulated in the progression of vascular aging, which contributes to vascular fibrosis and arterial stiffening [2], we further explored the effects of IMD on blood pressure and vascular fibrosis in the mouse vascular aging model. The results showed that heart weight-to-body weight ratio (HW/BW), systolic blood pressure (SBP), diastolic blood pressure (DBP), and pulse pressure (PP) were significantly increased in Adm2fl/flTagCre mice and in the Ang II-treated groups (Figure 3A–D), and their levels were more significant in Ang II-treated Adm2fl/flTagCre mice. Compared to the Adm2fl/fl mice, collagen deposition determined by Masson’s trichrome staining was significantly increased in the aortic media layer in Adm2fl/flTagCre mice and was further elevated in Adm2fl/flTagCre mice treated with Ang II (Figure 2E,F). Consistent with the above results tendency, the mRNA levels of Col1a1 and Col3a1 were also notably increased in the Adm2fl/flTagCre mice compared with the Adm2fl/fl mice, and their levels induced by Ang II in the aortas of Adm2fl/flTagCre mice were significantly increased compared with those of Adm2fl/fl mice (Figure 3G,H).
In addition, prosenescent factors such as interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), monocyte chemotactic protein-1 (MCP-1), and transforming growth factor-β (TGF-β) are upregulated in aging blood vessels and exert a crucial effect on the pathogenesis of vascular aging [6]. In the present study, real-time PCR results showed that the mRNA levels of Il-6, Tnf-α and Tgfb1 were markedly increased in the Adm2^fl/fl^TagCre mice compared with the Adm2^fl/fl^ mice, Ang II infusion further increased their levels (Il-6, Tnf-α, Mcp-1 and Tgfb1) in the aortas of Adm2^fl/fl^TagCre mice (Figure 3I–L). The above results showed that IMD deficiency in VSMCs or Ang II treatment significantly increased blood pressure and aggravated vascular fibrosis and prosenescent factor expression in arterial media, and IMD deficiency in VSMCs significantly promoted Ang II-induced changes, suggesting that IMD deficiency may aggravate vascular senescent phenotype transition and promote the elevation in blood pressure and myocardial hypertrophy.
2.4. IMD Alleviated DNA Damage in Ang II-Treated Aortas
DNA damage is one of the most important causes of vascular aging [6]. The inhibitory effect of IMD on Ang II-induced vascular senescent phenotype transition may be achieved by alleviating DNA damage. We explored the effects of IMD on DNA damage in the Ang II-induced mouse vascular senescent phenotype transition model. Both p-H2A histone family member X (S139) (γH2AX) and p53-binding protein 1 (53BP1) are widely used biomarkers of DNA damage [20,21]. Immunohistochemistry results showed that Ang II treatment significantly elevated the protein levels of 53BP1 and γH2AX in Adm2^fl/fl^ mice, and this effect was further strengthened in Adm2^fl/fl^TagCre mice (Figure 4A,B,D,E). Moreover, the increased protein levels of 53BP1 and γH2AX in the arteries of Adm2^fl/fl^TagCre mice were confirmed by Western blot (Figure 4G–I), suggesting that IMD deficiency aggravated DNA damage in mouse aortas. In addition, the protein expression of α-actin of VSMCs phenotype was decreased in Adm2^fl/fl^ mice, and this decrease was more significant in Adm2^fl/fl^TagCre mice after Ang II treatment (Figure 4C,F). This further indicated that the VSMCs in the arterial media underwent a phenotypic transformation.
The effect of IMD on DNA damage was further confirmed in Ang II-induced senescent VSMCs in vitro. Consistent with the results in vivo, protein levels of 53BP1 and γH2AX in the nucleus of senescent VSMCs were significantly higher than those in the control group and were decreased under IMD_1-53_ treatment (Figure 4H–K). Similarly, exogenous IMD_1-53_ decreased the protein levels of 53BP1 and γH2AX in Ang II-treated VSMCs (Figure 4L–N). The above results suggested that IMD_1-53_ effectively alleviated DNA damage in senescent VSMCs.
2.5. IMD Inhibited DNA Damage and the Senescent Phenotype Transition of VSMCs by Activating PARP1
Previous studies have found that IMD upregulates the activity of NAD^+^-dependent deacetylases and plays a protective role in CVDs [15], whereas PARP1 is also an NAD^+^-dependent ADP-ribose polymerase and plays an important role in recognizing DNA damage and initiating DNA damage repair [22]. Therefore, we further investigated whether IMD could inhibit DNA damage and thus reduce vascular senescent phenotype transition by regulating PARP1. Western blot results showed no significant effect of IMD on the protein level of PARP1 in senescent VSMC induced by Ang II (Figure 5A,B). However, compared with the control group, poly ADP-ribose (PAR) protein level in Ang II-treated VSMCs was significantly decreased, while IMD_1-53_ restored PAR level induced by Ang II (Figure 5C,D), suggesting that IMD_1-53_ can significantly increase PARP1 activity. PJ34, an inhibitor of PARP1, was used to explore whether IMD repaired DNA damage by activating PARP1. In Ang II-treated VSMCs, IMD_1-53_ administration significantly inhibited SA-β-gal activity (Figure 5E,F) and decreased the protein levels of p16 and p21, while PJ34 treatment blocked the above effects of IMD_1-53_ (Figure 5G,J,K). Consistently, IMD_1-53_ treatment significantly decreased the protein levels of γH2AX and 53BP1 in Ang II-treated VSMCs, which were blocked by PJ34 (Figure 5G–I). Therefore, these results suggested that IMD_1-53_ could inhibit DNA damage and reverse VSMC senescent phenotype transition by increasing the activity of PARP1.
2.6. IMD Increased PARP1 Activity by Activating NAMPT
PARP1 activity relies on nicotinamide adenine dinucleotide (NAD^+^), and the nicotinamide phosphoribosyl transferase (NAMPT)-mediated salvage pathway is a major source of NAD^+^ in VSMCs [23]. In Ang II-treated VSMCs, the ratio of NAD^+^/NADH was significantly reduced, which was reversed by IMD_1-53_ (Figure 6A). Next, the NAMPT inhibitor FK866 was used to explore whether the effect of IMD_1-53_ on NAD^+^ increase was dependent on NAMPT. The protein level of PAR represented the activity of PARP1, which was significantly decreased in the Ang II treatment group. IMD_1-53_ increased the PAR protein level in Ang II-treated VSMC, which was blocked by FK866 (Figure 6B,C), suggesting that NAMPT was involved in IMD_1-53_ mediated PAR production. In addition, IMD_1-53_ treatment significantly decreased the protein levels of γH2AX and 53BP1, which were blocked by FK866 (Figure 6D–F). In addition to DNA damage markers, Ang II-induced elevated protein levels of p16 and p21 were strikingly decreased by IMD_1-53_, but this effect was also blocked by FK866 (Figure 6D,G,H), suggesting that NAMPT was involved in the role of IMD in improving vascular senescent phenotype transition. Collectively, these results suggested that IMD may increase PARP1 activity by activating NAMPT.
3. Discussion
Despite intensive studies on the cardiovascular protective function of IMD, its role in the pathogenesis of senescent phenotype transition of VSMCs has not been investigated. In this study, we provided novel evidence that IMD reversed the senescent phenotype transition of VSMCs in tunica media of aorta. Ang II-treated mice showed vascular senescent phenotype transition and decreased IMD expression in tunica media. SMC-specific IMD-deficiency exacerbated senescent phenotype transition of VSMCs and further promoted Ang II-induced transition of VSMCs, while exogenous IMD_1-53_ significantly ameliorated Ang II-induced senescent phenotype transition of VSMCs. Mechanistically, IMD enhanced the activity of PARP1 by activating NAMPT in VSMCs and increasing the levels of NAD^+^ in VSMCs, alleviating DNA damage and then delaying VSMC senescent phenotype transition.
Stress-induced premature senescence is related to CVDs. Ang II, the most potent component of the renin–angiotensin system, plays a central role in cardiovascular aging. A previous study showed that the Ang II concentration increased with age in nonhuman primates [24]. Moreover, Ang II has been shown to induce premature senescence in VSMCs and aortas [17,25]. Therefore, we used Ang II to construct a mouse model with the senescent phenotype transition of VSMCs in tunica media of aorta in mice. Our results showed that Ang II treatment increased the protein levels of p16 and p21 and the activity of SA-β-gal in the aortas of mice, indicating that VSMCs transition from contractile phenotype to senescent phenotype was successfully constructed. Additionally, increased collagen deposition and prosenescent factor levels further confirmed the vascular senescent phenotype transition induced by Ang II. We also used Ang II to induce VSMCs senescent phenotype transition in vitro, and the SA-β-gal activity and the protein levels of p16 and p21 were significantly increased in Ang II-treated VSMCs compared to the control group, suggesting that Ang II successfully induced senescent phenotype transition of VSMCs. Previous studies showing that high concentrations of Ang II may lead to VSMC apoptosis were considered [26].
IMD is a cardiovascular polypeptide involving the maintenance of cardiovascular homeostasis, and it exerts a potential beneficial role in CVDs. Increasing evidence shows that IMD has a protective effect on vascular abnormalities, including vascular calcification, vascular inflammation, and abdominal aortic aneurysm which all involve the process of vascular senescence [14,15,16,27,28]. A previous study showed that IMD was downregulated in naturally aged rat aortas [15]. In this study, IMD mRNA level was significantly decreased in the aortas of mice with vascular aging induced by Ang II. In the Adm2^fl/fl^TagCre mice, we found that IMD deficiency in VSMCs promoted prosenescent factor expression (Il-6, Tnf-α and Tgfb1), inhibited the expression of VSMCs marker protein (α-actin), and aggravated vascular senescent phenotype transition, which might indicate that IMD deficiency in VSMCs aggravated the elevation in BP and vascular fibrosis by promoting vascular aging in tunica media of aorta. Moreover, exogenous IMD_1-53_ administration ameliorated VSMCs senescent phenotype transition. These results suggest that IMD in the aortas may play an important role in inhibiting vascular senescent phenotype transition and further alleviating aging.
DNA damage is a critical characteristic and mechanism of cellular senescence [29,30]. Nuclear and mitochondrial DNA damage genomes both have been found in aging mammals [31,32]. 53BP1 and γH2AX are two general markers of DNA damage. When DNA double-strand damage occurs, the S139 site of histone H2AX is phosphorylated to form γH2AX [33]. 53BP1 is an important regulatory molecule of DNA double-strand damage, which can rapidly aggregate to chromatin near the damage site [34,35]. Our data showed the increased protein levels of 53BP1 and γH2AX both in Ang II-treated aortas and VSMCs, which were further enhanced in IMD deficient mice, and were ameliorated by exogenous IMD_1-53_ treatment in vitro. These results suggest that IMD may alleviate vascular senescent phenotype transition in tunica media by alleviating DNA damage.
Poly ADP-ribosylation is an immediate repair response to mitigate DNA damage and is catalyzed primarily by PARP1 [36,37]. After PARP1 recognizes DNA damage, it catalyzes the formation of PAR, which plays an important role in recruiting subsequent effector molecules [22]. Thus, the PAR protein level reflects the PARP1 activity [38,39]. PARP1 mitigated replicative cell senescence by activating homologous recombinant double-strand rupture repair [40]. Therefore, we speculated that PARP1 might be involved in the beneficial effect of IMD on Ang II-induced vascular senescent phenotype transition. We found that the protein level of PARP1 was significantly increased in Ang II-treated aortas and VSMCs, which suggested a critical role of PARP1 in Ang II-induced vascular senescent phenotype transition, indicating that the compensatory increase in PARP1 may be in response to the DNA damage induced by Ang II. Although exogenous IMD_1-53_ did not significantly regulate the protein level of PARP1 in VSMCs, IMD might promote DNA repair by increasing PARP1 activity. As previously reported, Checkpoint kinase 2 (CHK2) increases PARP1 activity, promoting the repair of oxidative DNA damage without influencing PARP1 protein expression [41]. Our data showed that the protein level of PAR was significantly elevated in the IMD_1-53_ treatment group, suggesting that IMD might increase PARP1 activity. Furthermore, the protective effects of IMD_1-53_ against DNA damage and VSMC senescent phenotype transition were blocked by the PARP1 inhibitor PJ34. These results suggested that IMD might inhibit VSMCs senescent phenotype transition by enhancing PARP1 activity.
The activity of PARP1 to catalyze the formation of PAR is NAD^+^-dependent [36]. NAD^+^ is synthesized through two pathways, namely the de novo pathway from amino acids and the salvage pathway from nicotinamide, and the latter is the main source of NAD^+^ in mammalian cells [42]. NAMPT is the rate-limiting enzyme of the salvage pathway and regulates the intracellular NAD^+^ pool, participating in the regulation of NAD^+^-dependent enzyme activity [23]. Reducing the cellular NAD^+^ content with the NAMPT inhibitor FK866 can significantly reduce the activity of PARP1. Consistently, our data showed that the relative content of NAD^+^ in Ang II-treated VSMCs was significantly decreased, which was significantly increased after IMD_1-53_ administration. In addition, the NAMPT inhibitor FK866 successfully blocked the effect of IMD_1-53_ on enhancing PARP1 activity and alleviating DNA damage. These results suggested that IMD might activate PARP1 by enhancing the activity of NAMPT. However, the specific molecular mechanism by which IMD regulates NAMPT warrants further exploration.
Previous studies demonstrated that Ang Ⅱ-induced hypertension and myocardial hypertrophy in mice could be improved by systemic IMD_1-53_ treatment [43]. Our present study indirectly showed that IMD located in the medial layer of the artery might have the ability to exert the anti-hypertensive effect through inhibiting Ang Ⅱ-induced remodeling of the arterial media. Mechanistically, the IMD loss of VSMCs might promote the transformation of the aging phenotype of VSMCs that further contribute to vascular remodeling, thereby increasing blood pressure and myocardial hypertrophy. One limitation of the current study is the absence of functional data in assessing vascular reactivity. For instance, the measurements of acetylcholine-induced vasodilation would strengthen the evaluation of endothelial cell function, which can further support the protective role of IMD in vascular tissue. Another limitation is that only using male animals for exploring the effect of IMD on the vascular senescent phenotype transition is insufficient. Therefore, we will pay attention to these limitations and improve the relevant experiment in future research.
In conclusion, our study discovered a novel mechanism that IMD can improve vascular senescent phenotype transition in tunica media by alleviating DNA damage. Mechanistically, IMD enhanced PARP1 activity by increasing NAMPT-mediated NAD^+^ production, resulting in the promotion of DNA damage repair and improvement in VSMCs senescent phenotype transition. This study uncovered the beneficial effect of an endogenous active peptide on vascular senescent phenotype transition in tunica media, which may provide a new target for the prevention and therapy of vascular aging-related diseases.
4. Materials and Methods
4.1. Materials
Antibodies for IMD (Sc-86272), β-actin (Sc-47778), and all secondary antibodies (horseradish peroxidase-conjugated anti-mouse, anti-rabbit, or anti-goat IgG) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for poly [ADP-ribose] polymer (PAR, Ab14459), p21 (Ab109199), p16INK4a (p16, Ab189034), 53BP1 (Ab36823), and p-H2A histone family member X (S139) (γH2AX, Ab26350) were from Abcam (Cambridge, UK). Antibodies for α-actin and PARP1 (CST9532) were from Cell Signaling Technology (Danvers, MA, USA). The nitrocellulose membrane was from Amersham Life Science (Amersham, UK). PJ34 (HY-13688A) was from MedChemExpress (Monmouth Junction, NJ, USA). FK866 (A4381) was from Apexbio (Houston, TX, USA). The sequences of oligonucleotide primers for Real-time PCR amplification were synthesized by Tsingke Biotechnology (Beijing, China). TRIzol reagent and the enhanced chemiluminescence (ECL) kit were from Applygen Technologies (Beijing, China). The SuperReal PreMix Plus (SYBR Green) kit and The FastKing RT Kit (with gDNase) for Real-time PCR were from Tiangen Biotech (Beijing, China). Alzet mini-osmotic pumps were from DURECT Corp (model 1004, Cupertino, CA, USA). Synthetic human IMD_1-53_ and human angiotensin II (Ang II) were from Phoenix Pharmaceuticals (Belmont, MA, USA). Other chemicals and reagents were of analytical grade.
4.2. Animals
All animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011) and passed verification by the Animal Care Committee of Peking University Health Science Center. Adm2^fl/fl^TagCre mice were generated on a C57BL/6 background from the Animal Research Center of Nanjing University (Cat. NO.131110, Nanjing, China) and were previously generated by crossing mice carrying loxP-flanked Adm2 alleles with Tagln-Cre transgenic mice (C57BL/6, Cat. NO. NM-KI-200144, Shanghai, China). Mice (male) were randomly assigned to 4 groups (n = 10 each group): Adm2^fl/fl^ Saline, Adm2^fl/fl^TagCre Saline, Adm2^fl/fl^ Ang II, and Adm2^fl/fl^TagCre Ang II. For the vascular aging model, 12- to 14-week-old male Adm2^fl/fl^TagCre mice and their age-matched Adm2^fl/fl^ mice underwent the application of saline or Ang II (dissolved in saline) by subcutaneous implantation of an Alzet osmotic minipump. Saline or Ang II (1.44 mg/kg/d) were then continuously infused for 4 weeks [17]. All animals were fed normal mouse chow. After 4 weeks, blood pressure was measured in conscious mice by the standard tail-cuff methods as described [18]. At the end of the experiment, mice were anesthetized by using 3% isoflurane administration via inhalation through a mask. After anesthetization, all mice were killed by exsanguination, and the aortas were quickly removed for further analysis.
4.3. Senescence-Associated β-Galactosidase Staining
Senescence-associated β-galactosidase (SA-β-gal) staining was performed using a commercial kit (#9860S, Cell Signaling Technology, Danvers, MA, USA). Segments of mouse thoracic aortas were freshly placed in optimal cutting temperature compound and cut into 7-μm-thick sections. Aorta sections or VSMCs were fixed by using 4% paraformaldehyde for 15–20 min, washed with PBS, incubated in β-galactosidase staining solution at 37 °C for 12 h, and then Eosin re-staining was conducted and photographed. Staining data were quantified by using Image-Pro Plus v6.0.
4.4. Masson’s Trichrome Staining
Segments of mouse aorta (1 cm) were placed in 4% phosphate buffered neutral formalin (pH 7.4, 0.1 mol/L) for 12 h and then immersed in 20% sucrose solution for storage. Aorta samples were dehydrated and embedded in paraffin, cut into 5-μm-thick sections, and then subjected to Masson’s trichrome staining (G6004, Servicebio, Wuhan, China). For Masson’s trichrome staining, the arterial media was delineated and the percentage of media positive for blue staining was assessed by using Image-Pro Plus v6.0 (Media Cybernetics, Rockville, MD, USA).
4.5. Immunostaining
Segments of mouse thoracic aortas embedded in paraffin underwent immunohistochemical staining. Sections (5 μm) were incubated with antibodies against IMD (1:40), 53BP1 (1:500), γH2AX (1:100), or α-actin (1:200) at 4 °C overnight, and then with secondary antibody for 1 h at 37 °C. Nuclei were stained with hematoxylin and then treated with diaminobenzidine. Immunostaining data were quantified by using Image-Pro Plus v6.0. VSMCs were first fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.3% Triton X-100 for 10 min. Nonspecific binding was reduced by incubation in 1% bovine serum albumin diluted in PBS for 60 min at 37 °C. VSMCs were incubated with antibodies against 53BP1 (1:200) or γH2AX (1:100) at 4 °C overnight, rinsed with PBS and incubated with fluorescein-labeled secondary antibodies. Nuclei were stained with Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA). Images were acquired under fluorescence microscopy (Leica, Cambridge, UK).
4.6. Real-Time (RT) PCR Analysis
Total RNA from aortic tissues was isolated and reverse transcribed. RT-PCR amplification was performed using the Applied Biosystems 7500 fast PCR System (Waltham, MA, USA) and SYBR Green I reagent (#FP205-02, Tiangen Biotech, Beijing, China). The cycle threshold (Ct) was determined as the number of PCR cycles required for a given reaction to reach an arbitrary fluorescence value within the linear amplification range. Relative quantification was performed according to the 2^−δΔCt^ method with the GAPDH level as a reference. The forward and reverse PCR primers sequences and annealing temperature were listed as follows in Table 1.
4.7. Western Blot Analysis
Extracts of aortic tissues or cells containing equal amounts of total protein were resolved by 10% or 12% SDS-PAGE and then transferred to a nitrocellulose membrane. Nonspecific proteins were blocked with 5% nonfat dried milk for 1 h, and then incubated with primary antibodies for β-actin (1:3000), 53BP1 (1:5000), γH2AX (1:500), p21 (1:500), p16 (1:500), PARP1 (1:1000), and PAR (1:1000) overnight at 4 °C. The secondary antibody (1:2000) was incubated for 1 h at room temperature. The labeling of proteins was shown by enhanced chemiluminescence. The amounts of proteins were analyzed by using NIH ImageJ (v1.8.0) and normalized to β-actin expression.
4.8. Cell Culture
Male mouse VSMCs were isolated from 8-week-old C57BL/6J mice as described in [14]. In brief, thoracic aortas were digested by using 1 mg/mL type II collagenase (#LS004176, Worthington Biochemical Corp, Lakewood, NJ, USA) at 37 °C for 10–15 min. The adventitia and endothelium were removed. Aortas were placed in DMEM containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin overnight in a 37 °C incubator with 5% CO_2_, and then digested in 5 mL of 1 mg/mL type II collagenase and 0.25 mg/mL elastase I (#E1250, Sigma-Aldrich, St. Louis, MO, USA) at 37 °C for 90 min. After centrifugalization at 1000 rpm for 3 min, the cells were cultured with DMEM containing 20% FBS with 5% CO_2_ at 37 °C. VSMCs at passage 5–7 were used for the relevant experiments. As previously described in [14], confluent VSMCs were incubated in DMEM containing 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin, treated with Ang II (10^−7^ mol/L) or IMD_1-53_ (10^−7^ mol/L), and cultured at 37 °C in an incubator containing 5% CO_2_ and 95% air. After treatment for 3 days, the cells were collected for the relevant experiments. To explore the signaling pathway by which IMD_1-53_ involves VSMC senescence, VSMCs were preincubated with the PARP1 inhibitor PJ34 (5 μmol/L) or the NAMPT inhibitor FK866 (5 μmol/L) for 30 min.
4.9. Intracellular NAD+/NADH Content
Total intracellular NAD^+^/NADH was assessed using the NAD^+^/NADH detection kit (S0175, Beyotime, Shanghai, China) referring to the manufacturer’s instructions. VSMCs were collected and directly lysed in lysis buffer from the detection kit. Total NAD and NADH in the extract were detected by colorimetry with a microplate reader (iMARK, BioRad, Hercules, CA, USA).
4.10. Statistical Analysis
All data are expressed as mean ± SD and were analyzed by GraphPad Prism 9.20 (GraphPad Software Inc., San Diego, CA, USA). The Kolmogorov–Smirnov test was used to evaluate the normality of the data distribution (p > 0.1), and the F-test was used to compare variances (p > 1 take for equal variance). All data showed normal distribution and passed equal variance testing. When dealing with small sample sizes (n = 4), Mann–Whitney test for the analysis of two groups or the Kruskal–Wallis test for comparing more than two independent groups were used. When dealing with sample sizes (n = 4), the unpaired Student’s t-test was used to identify significant differences between the two groups (n > 4). Comparisons of more than two groups in cellular experiment were analyzed by one-way analysis of variance (ANOVA, n > 4). Differences among the different animal groups were evaluated using two-way ANOVA (n > 4). When significant differences were detected by ANOVA, Tukey’s test was used for multiple comparisons. Statistical significance was accepted at p < 0.05.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rahman M. Siddik A.B. Anatomy, Arterioles Stat Pearls Stat Pearls Publishing Treasure Island, FL, USA 202332310381 · pubmed ↗
- 2Lacolley P. Regnault V. Segers P. Laurent S. Vascular smooth muscle cells and arterial stiffening: Relevance in development, aging, and disease Physiol. Rev.2017971555161710.1152/physrev.00003.201728954852 · doi ↗ · pubmed ↗
- 3Lacolley P. Regnault V. Avolio A.P. Smooth muscle cell and arterial aging: Basic and clinical aspects Cardiovasc. Res.201811451352810.1093/cvr/cvy 00929514201 · doi ↗ · pubmed ↗
- 4Lacolley P. Regnault V. Nicoletti A. Li Z. Michel J.-B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles Cardiovasc. Res.20129519420410.1093/cvr/cvs 13522467316 · doi ↗ · pubmed ↗
- 5Gan L. Liu D. Liu J. Chen E. Chen C. Liu L. Hu H. Guan X. Ma W. Zhang Y. CD 38 deficiency alleviates Ang II-induced vascular remodeling by inhibiting small extracellular vesicle-mediated vascular smooth muscle cell senescence in mice Signal Transduct. Target. Ther.2021622310.1038/s 41392-021-00625-034112762 PMC 8192533 · doi ↗ · pubmed ↗
- 6Ungvari Z. Tarantini S. Donato A.J. Galvan V. Csiszar A. Mechanisms of vascular aging Circ. Res.201812384986710.1161/CIRCRESAHA.118.31137830355080 PMC 6248882 · doi ↗ · pubmed ↗
- 7Ou H.-L. Schumacher B. DNA damage responses and p 53 in the aging process Blood 201813148849510.1182/blood-2017-07-74639629141944 PMC 6839964 · doi ↗ · pubmed ↗
- 8Jackson S.P. Bartek J. The DNA-damage response in human biology and disease Nature 20094611071107810.1038/nature 0846719847258 PMC 2906700 · doi ↗ · pubmed ↗